Hello,
I am trying to calculate the potential mean force of a ethanol molecule to enter a polymer membrane.The system has ethanol and water mixture and PIM-1 polymer membrane. I did the minimization , swelling simulation ( nvt with 1 bar pressure) for 10 ns, after that 2ns Npt simulation.All the simulation ran perfectly. Then I am trying to calculate the pmf in NVT simulation with using the colvars. I initially run for 1 ns and with 5.00 wall constant ( If I increase more than 5 the wall constant then the simulation have error of bond missing).
However, I am not getting the typical PMF profile as expected. I have four window , for each window the ethanol molecule is different ( but as they are identical I assumed that this doesn’t create any problem). After the merging of all the window, I have the below profile. The value for win0 and starting point of win1 should be close to zero as the ethanol is in bulk reservoir there but it is showing the opposite trend of value.
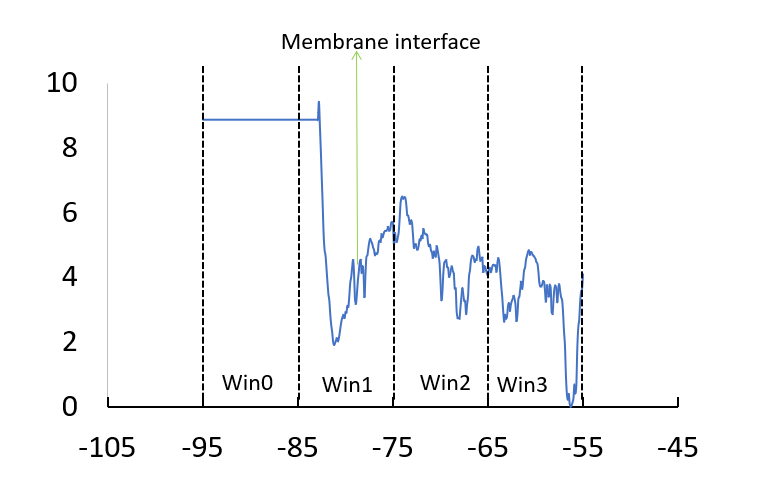
Can anyone suggest what might be the problem ?
----Input Scirpt----
System------------------------------------------------------------------------------------
units real
atom_style full
dimension 3
newton on
boundary p p p
timestep 1.0
Styles--------------------------------------------------------------------------------------
read_data NPT_2ns.data
bond_style hybrid harmonic
angle_style hybrid harmonic
dihedral_style hybrid harmonic opls
improper_style none
pair_style hybrid lj/charmm/coul/long 9.0 10.0 10.0 tersoff
include System_ethanol_water_coeff.in
special_bonds amber
kspace_style pppm 0.00001
#Define group and region -------------------------------------------------------------------
#spearate the polymer & water group
group wall type 12
group ethanol type 13 14 15 16 17 19
group water type 18 20
group polymer type 1 2 3 4 5 6 7 8 9 10 11
group wall1 id 6791:7934
group wall2 id 7935:9078
region membrane block -2.8255 51.1745 -1.558001 52.441999 -64.52 -6.52 units box
region left_block block -2.8255 51.1745 -1.558001 52.441999 -118 -88 units box
region right_block block -2.8255 51.1745 -1.558001 52.441999 10 40 units box
group Left_block region left_block
group Right_block region right_block
group Membrane region membrane
group waterinmembrane intersect water Membrane
group ethanolinmembrane intersect ethanol Membrane
group bkwater union Left_block Right_block water
group bkethanol union Left_block Right_block ethanol
#system_specific_setting-------------------------------------------------------------------------
fix fshake water shake 0.000001 2000 0 b 22 a 36
fix makestable wall setforce 0.0 0.0 0.0
#fix polymerstable polymer setforce NULL NULL 0.0
fix spring_polymer polymer spring/self 0.000239
fix 1 all nvt temp 300.0 300.0 100.0
fix Colvars all colvars ABF.colvars.inp seed 2122
thermo_style custom step temp vol
thermo 1000
run 1000000
—Colvars Scipt example for window 2—
colvarsTrajFrequency 1000 # output every 10 steps
colvarsRestartFrequency 1000
colvar {
name dist
width 0.1 # Spacing of grids
lowerBoundary -75.0
upperBoundary -65.0 # Determined by size of unit cell used
lowerWall -75
upperWall -65
lowerWallConstant 5.0 # Change this value as needed
upperWallConstant 5.0 # Change this value as needed
#outputAppliedForce on # keep track of bias force on this variable
distanceZ {
main {
atomNumbersRange {20560-20568}
}
ref {
atomNumbersRange {1-6790}
}
axis (0.0, 0.0, 1.0)
}
}
abf {
colvars dist
fullsamples 1000
Please suggest which part might be a mistake here.
Best Regards,
Morshed Mahmud