Hello, I am trying to simulate polymer degradation (i.e. breaking of chains or clusters) due to high temperature using ReaxFF force field in LAMMPS. I am using a relatively newer set of ReaxFF parameters (2020), which are capable to be used for high temperature experiments. The system was polymerized after subjecting a set of monomers to fix bond/create command and introducing dummy atom types for joining monomers.
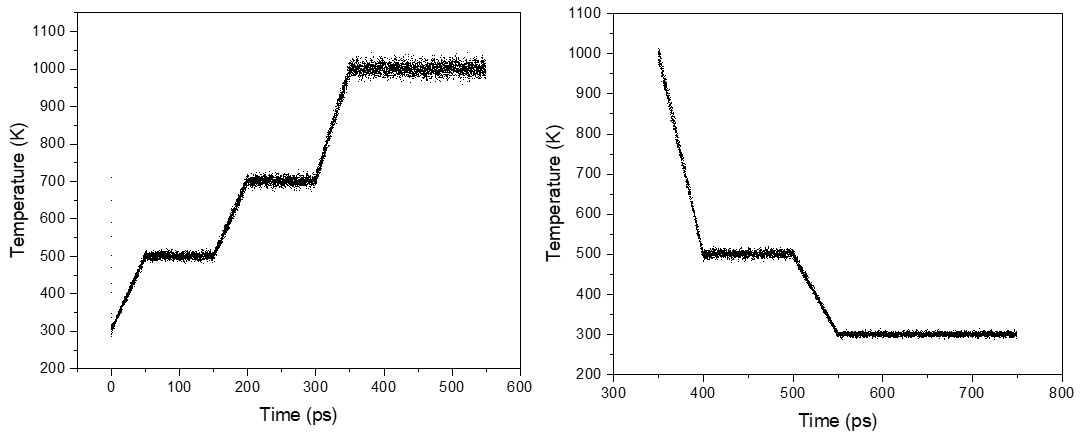
The system (4250 atoms or 21 chains in a box) which is initially kept at room temperature, is then subjected to high temperature (1000K) and then brought back to room temperature. (image below)
For the polymer system I am working with, the existing clusters (independent molecules) should breakdown into smaller fractions because of broken bonds at high temperature. i.e. smaller fractions should gradually increase, larger fractions should gradually decrease. However, I observed some strange behavior contrary to my expectation:
- The number of cluster goes down with progress of simulation:
Moreover, the fractions are multiples of the monomer, signaling that inter-monomer linkage is the weakest link in the chain segment and not any bond within the monomer. (As per chemistry for my polymer, the bonds within the polymer should also break at high temperature) However, no fractions present were fragments of monomer, but all of the fractions were multiples of monomers.
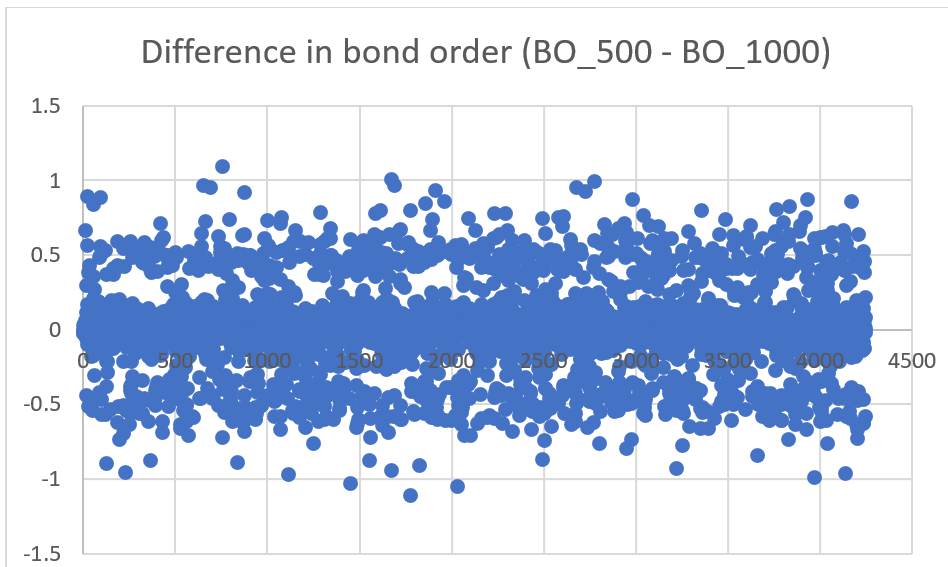
- Bond orders (BO) of all the bonds present in the system were logged and observed at two different temperatures 500K & 1000K. (Timestep 510000 and 5500000 respectively) It was expected that at higher temperature bond order would reduce because of weakened bonds (longer bond length). However, the bond order difference i.e. BO_500K – BO_1000K when plotted came out to be a curve with random distribution of points:
Can I get the system to work as per the chemistry of the polymer at high temperature? Any thoughts would be appreciated.
I am confused by this statement. ReaxFF does not use explicit bonds which is what fix bond/create will generate. Using a bond topology with ReaxFF will result in a bogus simulation unless precautions are taken. So can you please elaborate?
Sorry… this needs clarification. The fixed-bond OPLS all-atom force field was used for the initial set-up of the model. The box was densified using the ‘fix deform’ LAMMPS command.
Simulated polymerization took place immediately after densification, where bonds were created between the atoms at the ends of monomers using the ‘fix bond/create’ command.
Once the MD models were fully polymerized, the force field was changed from fixed-bond to
ReaxFF for the aging experiment.
That does not say whether you have changed the atom style and/or removed the topology information and/or changed the special_bonds setting. Only changing the various force styles is not sufficient.
Even though the original code given to me used atom style molecular and special_bonds extra 40 command, I changed the atom style to full and commented out the special bonds command because I was getting errors with them. Was this what’s wrong?
While in ReaxFF, I used atom style full.
I would have to see the files to make a proper assessment. But atom style molecular sounds wrong for OPLS/AA even since it requires partial charges, but charges (and bonds) would require atom style full.
ReaxFF calculations usually are done with atom style charge (which contains support for charges but not for bonds). There is no “special_bonds extra 40” command.
All of that combined hints at that you are not as experienced with the technical details for how to set up such a calculation than you probably should be (or else you would have been able to give me more specific and different answers). This doesn’t say anything about whether your calculations were correct or not, but there is a finite probability, but I need to see all the details to be certain.
Please find the necessary files in this github folder:
Thanks.
The ReaxFF calculations in these folders must be ok because they use atom style charge, so there cannot be any bond topology data that would cause problems or require additional settings to negate them.
However, you said you used atom style full and I don’t see anything like that here.
Also, you said that you had had errors and made other modifications. So I would need to see the real files for your runs including the data files.
The “OPLS-AA” calculations are either not really using OPLS-AA or are incorrect because they are missing the partial charges, since they use atom style molecular but should be using full. OPLS-AA has an increment system which must be used to compute the partial charges on hydrogen and carbon and other atoms. See for an example with a few simple molecules here: Homepage of Axel Kohlmeyer - Tutorial - Part 2
However, since those calculations are only used to prepare the system, they may still be acceptable, since the production was done with ReaxFF and if there was sufficient equilibration time the ReaxFF run may have corrected for geometry errors.
Good to know that the original calculations are correct.
The only changes I made were that I changed the atom style while in OPLS to full and commented out the special bonds command. And I switched from charge to full atom style when I was using ReaxFF forcefield. I found that only on doing so, the codes would run without any error. Please find my modified versions of the same files along with data files in this folder:
https://drive.google.com/drive/folders/1-E0_P84sODFb-RB3UVHTigPNhh82lihn?usp=sharing
The sequence is densification → polymerization → transition to reaxff → equilibration in reaxff.
During polymerization stage, monomers were bonded together using fix bond/create command. This command was used to form bonds between two dummy ‘reactive’ atoms at the two ends of the monomer. (atom types 6 & 7) Has it been done correctly?
From what I can see, in your simulations the OPLS-AA part has been done correctly in this case as far as the atom style and force field settings are concerned.
The conversion to ReaxFF and the ReaxFF setup also look correct. The choice of timestep is careful. The bond topology is removed as it should and despite the different atom style, the data file is correct and suitable and the included structure looks very reasonable on visualization.
However, there is one potential big problem here:
fix 3 all reax/c/species 100 100 10000 species.reaxc.PEEK_amorphous
This command will postpone all neighborlist updates so they will only be done every 10000 timesteps. That may be too infrequent, especially at high temperatures, and thus your force computations are missing significant contributions because atoms have moved around. You need to check your log files for “Dangerous builds”. If this is a significant number (> 10), then your simulations are not very good. Please see the “Warning” box on the page for the fix reaxff/species command — LAMMPS documentation (the name has changed for newer versions of LAMMPS, but the functionality is unchanged).
Yes I will be careful about the fix reax/c/species command and will also check for any dangerous builds. However, what do you think about the way it is behaving at high temperature? (contrary to chemistry of the polymer) That,
-
The number of cluster goes down with progress of simulation. Moreover, the fractions are multiples of the monomer, signaling that inter-monomer linkage is the weakest link in the chain segment and not any bond within the monomer. This polymer follows the random chain scission mechanism where ketone, ether groups break first. (around 700K), but I didn’t find any evidence of that.
-
At higher temperature, bond order is expected to reduce because of weakened bonds However, the bond order difference (values obtained from Reaxff file) when plotted came out to be a curve with random distribution of points:
Sorry, I have no comment on that. This is beyond anything that I have done research on and I don’t want to speculate. I can really only discuss “technical matters”.
Thank you for the help. Will come back here in case of questions.