I am simulating a polymer of 414 beads in NVT ensemble. The polymer has rigid bodies that are integrated with the rigid/nvt thermostat. These are the lines I use:
group CD1 id 2:38rigid body group nonrigid subtract all CD1#flexible parts
The simulation runs normally. However, I get a weird fluctuation in the stress tensor in the pxx component printed in the log file. Here I show a plot of pxx (black), pyy (red), and pzz (green):
To me, it does not make any sense that one of the components of the stress tensor has higher deviation since the system is homogenous and isotropic averaged over time, but the fluctuations remain in the x coordinate. I understand that this could happen before equilibration, but I have run this simulation for over 100e6 timesteps and I still get the same result. Any idea of what could be happening?
The LAMMPS version I’m using is the Stable releas of 2 Aug 2023.
Please see the forum guidelines for properly quoting from text files in the forum. Your quotes are garbled and partially misformatted.
There is no preference for any direction in the source code, so this makes it unlikely that there is a bug. The next best explanation would be the specifics of your system, i.e. the number and shape of your rigid objects versus the box. “414 beads” doesn’t sound that you have a large system, and there may be entanglement that cannot easily resolve. What happens if you swap the column for y and z coordinates? or simulate a larger system with 2, 4, or 10 times as many beads?
Please note that with small systems and small boxes, you can have unexpected artifacts from non-spherical objects due to inter-acting with atoms of the same object due to periodic boundaries depending on orientation.
Please check that your system is in fact homogeneous and isotropic over time. You can do so by comparing the x-, y- and z- co-ordinate mean squared displacements over time, or even just their standard deviations over time.
If the thermostatted parts of your system have much less mass than the rigidified components, those parts may not have enough “thermalizing power” to homogenise your system’s density and orientation fast enough.
You might also be interested in the compute gyration command. You can compare all the components of the gyration tensor and compare the extension of your polymer in the 3 direction of space. You can also use gyration/shape to see how far your molecule is from ideal isotropic case.
Also check that there is no remnant momentum in your initial velocity distribution, and you might consider using the keyword zero in your langevin fix to avoid other drift from the random forces on long runs.
This is a good idea indeed, I’ll try this. However I still don’t understand how this inhomogeneity in the stress tensor is sustained after these many timesteps.
The box is a 200^3 square as I stated above.
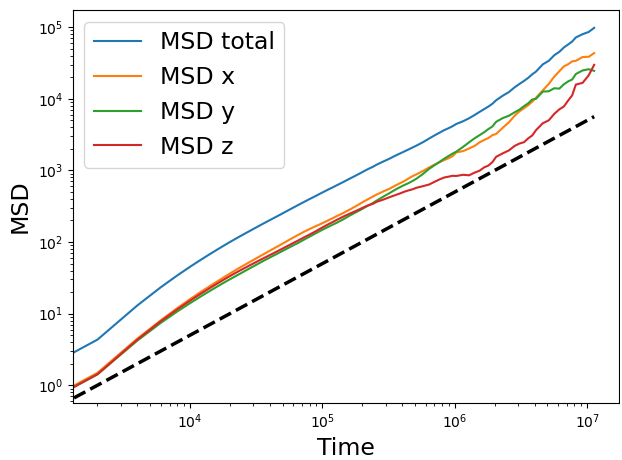
I have calculated the MSD of the polymer center of mass and got a fairly homogeneous result for the three components considering the noise at long times:
The black dashed line shows the diffusive regime.
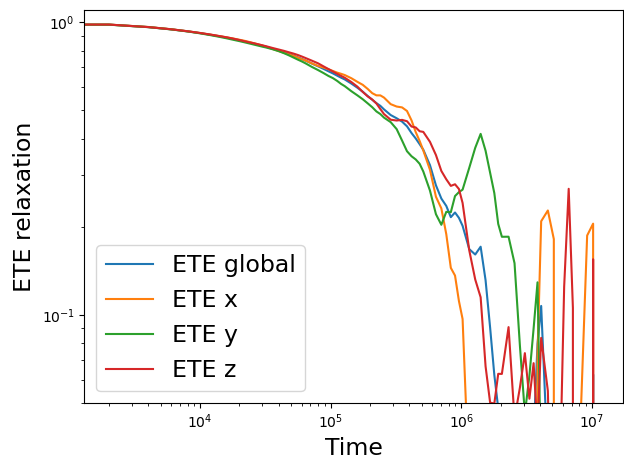
I also calculated the end-to-end (ETE) relaxation and the three components to check the reorentation dynamics and it seems there is not preference:
The end-to-end is normalized by the initial value.
I have also simulated different parts of the polymer alone and only two of them show the anisotropy in the stress tensor containing rigid bodies. Still the MSD was homogeneous
Hi, so no reply on this thread for a while. Do you think this anomaly may be a bug in calculating the stress tensor when dealing with rigid bodies? Otherwise, what else can I do? In my opinion if there is any entanglement in the system the stress should be homogeneous thtoughout the three components of the stress tensor, as the ETE relaxation demonstrates reorentation.
Difficult to say based on the available information. You apparently haven’t been successful at convincing anybody so far.
The gap between having data that looks suspicious and convincing proof of a bug can be large and difficult to cross. That is not unusual in research. Since you are concerned about functionality that has been around a long time and was used by a large number of people, it is quite possible that the implementation is correct. On the other hand it is also plausible that your system has some very unusual properties and those trigger an unexpected use case for which the implementation may have a problem. Over the years, I have seen both, but the proof that some functionality that has been used a lot has a flaw is particularly difficult since it has to be convincing beyond any doubt.
I have no specific suggestions for how to proceed. Since not all LAMMPS developers monitor the forum, you could consider submitting a bug report issue on GitHub.
As Axel says a bug in the computation of one element of the pressure tensor is unlikely given the state of the code. It is not like they are computed one after another independently. This does not leave many leads concerning this. There can be either an issue concerning the forces or velocities isotropically which (is also unlikely but not impossible) or this is the physics of your model.
Maybe try simulating a 90 degrees rotated molecule with it larger axis along another direction to see if you have the same “weird fluctuation” along the x axis. And if yes maybe there is a bug, but that would be up to you to show why it shows up in your system or provide enough resources for people to track it down.
EDIT: I couldn’t help myself but checked the values a simple Kremer-Grest polymer model (reduced units, no attractive interaction beside bonds) gave me with the few elements you gave from your input. I saw no differences in fluctuation in the pressure tensor (all three variances \approx{}2.16\cdot{}10^{-7}, all average values close to 2\cdot10^{-7}). I see no reason to believe there is a bug here.
Here are my relevant input files for comparison. data.lmp (40.8 KB) in.lmp (1.0 KB)