Dear Lammps Users,
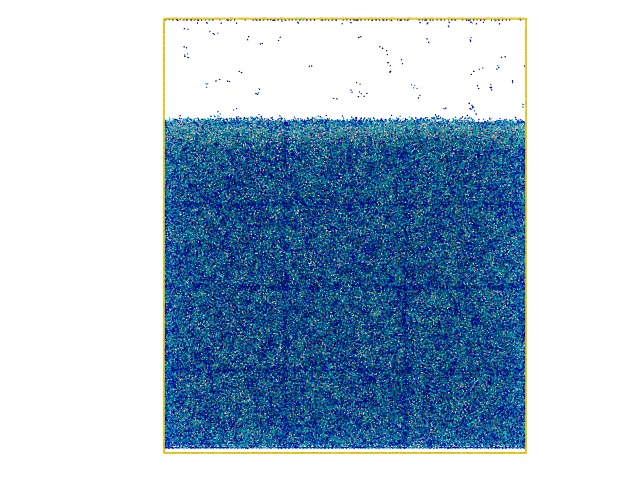
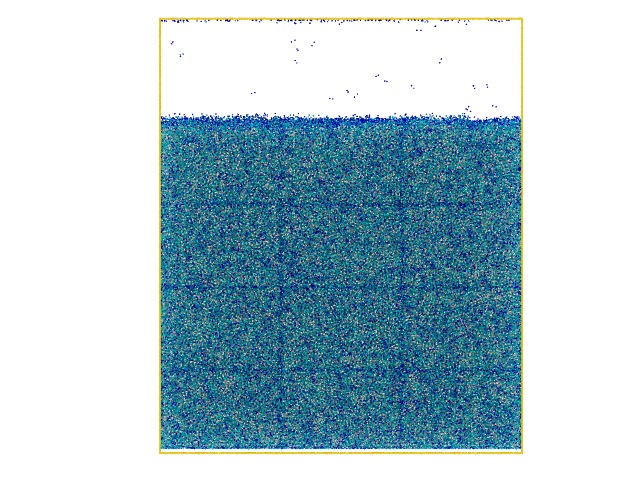
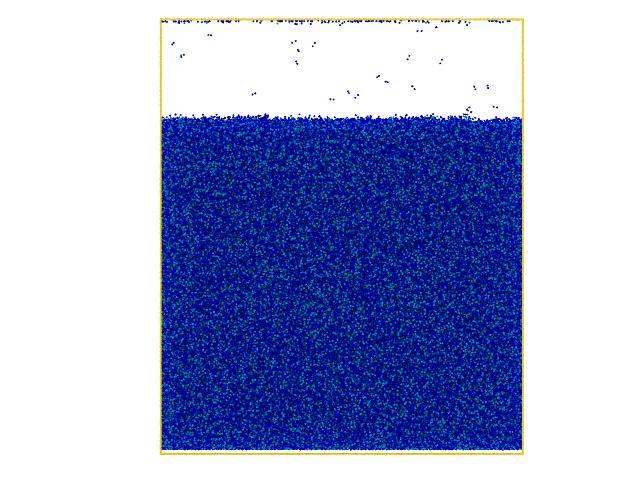
I recently built up a gel system with LJ beads and let them crosslink using fix bond/create to create a highly crosslinked polymer gel. However I found an interesting result in which the simulation box is divided into an lattice structure and the total cell number is exactly equal to the number of CPUs I use (please check the attached snapshots).
The running command I use is:
mpirun -np 36 lmp_ompi_g++ -sf gpu -pk gpu 4 -in system.in
My input script is:
----------------- Init Section -----------------
units lj
atom_style full
pair_style lj/cut 2.5
pair_modify shift yes
bond_style harmonic
boundary p p p
timestep 0.001
neighbor 2.5 bin
neigh_modify every 1 one 10000
special_bonds lj 0 1 1 extra 50
comm_modify cutoff 5
----------------- Atom Definition Section -----------------
read_data “system.data”
pair_coeff * * 1.5 1.0 2.5 #epsilon sigma cutoff 1KT KJ/mol in unit
bond_coeff * 30 1.0
----------------- Run Section -----------------
equalibrium
group monomer type 1 2 3 4 5 6
group ms type 1 2 3 4 5 6
fix 1 ms nve
fix 2 ms langevin 1.0 1.0 10.0 1234 #KT=2910
thermo 1
fix bl all balance 1000 1.1 shift z 100 1.1
min_style fire
minimize 1.0e-4 1.0e-6 1000 1000
timestep 0.005
reset_timestep 0
#M1 + M1 = M2 + M2
fix p1 all bond/create 1 1 1 1.12246 1 iparam 1 2 jparam 1 2 prob 1 2
#M2 + M1 = M3 + M2
fix p2 all bond/create 1 2 1 1.12246 1 iparam 1 3 jparam 1 2 prob 1 3
#M3 + M1 = M4 + M2
fix p3 all bond/create 1 3 1 1.12246 1 iparam 1 4 jparam 1 2 prob 1 4
#M4 + M1 = M5 + M2
fix p4 all bond/create 1 4 1 1.12246 1 iparam 1 5 jparam 1 2 prob 1 5
#====M2 + M2 = M3 + M3
fix p5 all bond/create 1 2 2 1.12246 1 iparam 1 3 jparam 1 3 prob 1 6
#M3 + M2 = M4 + M3
fix p6 all bond/create 1 3 2 1.12246 1 iparam 1 4 jparam 1 3 prob 1 7
#M4 + M2 = M5 + M3
fix p7 all bond/create 1 4 2 1.12246 1 iparam 1 5 jparam 1 3 prob 1 8
#====M3 + M3 = M4 + M4
fix p8 all bond/create 1 3 3 1.12246 1 iparam 1 4 jparam 1 4 prob 1 9
#M4 + M3 = M5 + M4
fix p9 all bond/create 1 4 3 1.12246 1 iparam 1 5 jparam 1 4 prob 1 10
#====M4 + M4 = M5 + M5
fix p10 all bond/create 1 4 4 1.12246 1 iparam 1 5 jparam 1 5 prob 1 11
dump myDump all atom 1 dump.atom
run 2
write_data final.data.*