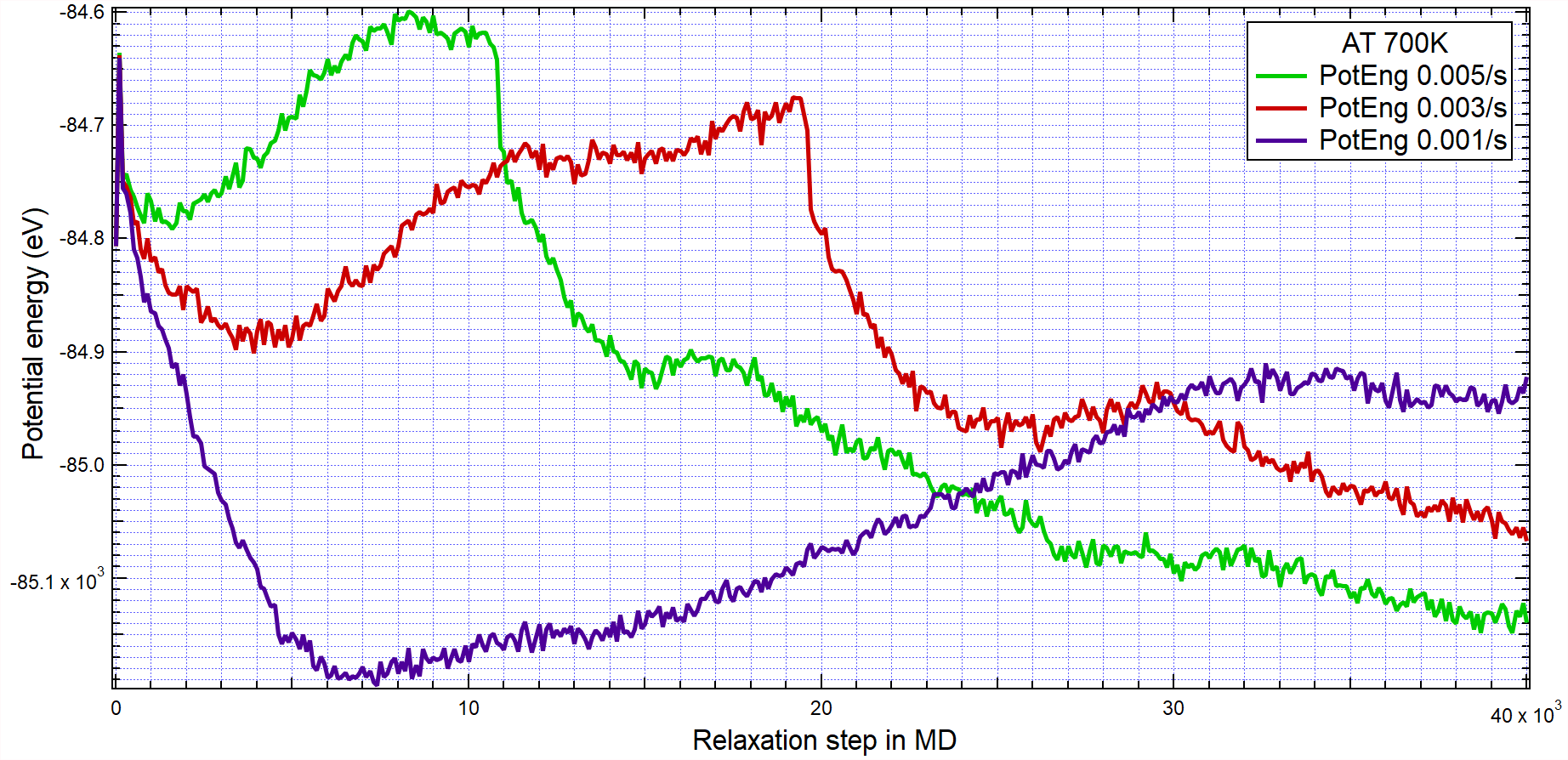
Dear all Lammps users,
Why does the potential energy curve increase first, then decrease with growing steps in MD simulation?
Why should it not?