Hello,
I am trying to simulate a Copper-Coated SWCNTs in Al Metal Matrix Composites to find its mechanical characteristics. Before Minimization and Thermal Equilibration, I have frozen cnt and coating. When I am running my code, the matrix is getting distorted.
Before:

After:
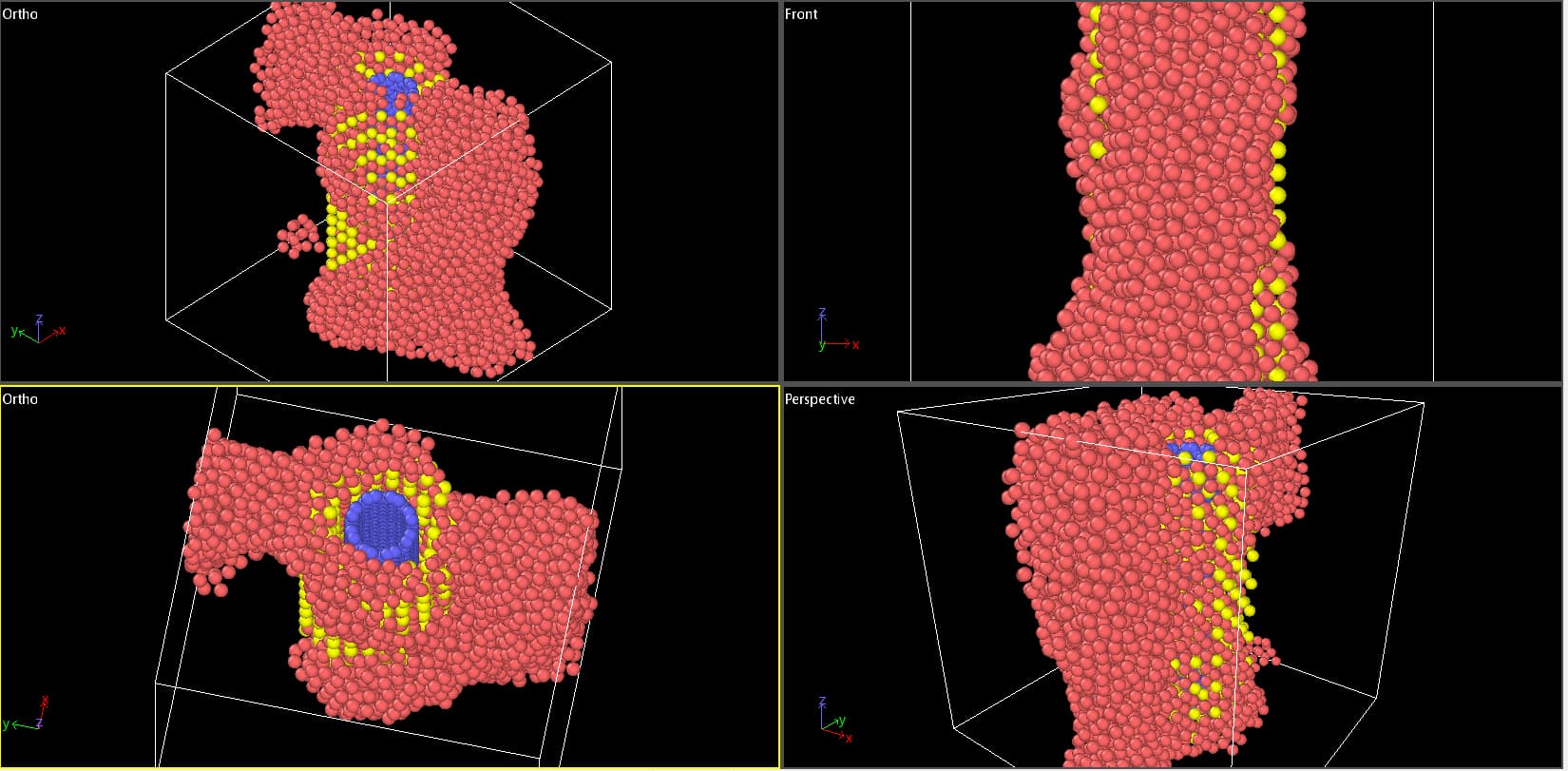
Is there any problem with defining Interatomic Potential ?
What should I do now?
Code:
units metal
variable T equal 300
variable dt equal 0.01
dimension 3
boundary p p p
atom_style atomic
read_data model.lmp
#### Assign atom types
mass 1 26.98 # Al
mass 2 12.01 # C
mass 3 63.546 # Cu
#### Define interatomic potentials using hybrid
pair_style hybrid airebo 10.2 eam/alloy lj/cut 4.5
pair_coeff * * airebo CH.airebo NULL C NULL
pair_coeff 1 2 lj/cut 0.03438 3.01
pair_coeff 3 2 lj/cut 0.02578 3.0825
pair_coeff * * eam/alloy al-cu-set.eam.alloy Al NULL Cu
#### Define groups
group CNT type 2
group coating type 3
group CNT_coating union CNT coating
group matrix subtract all CNT_coating
#### Freeze CNT and coating atoms during minimization
fix freezeCNT_coating CNT_coating setforce 0.0 0.0 0.0
#### Define simulation parameters
neighbor 2 bin
neigh_modify delay 0 every 1 check yes
#### energy minimization
min_style cg
minimize 1.0e-6 1 10000 10000 # Energy tolerance = 1.0e-6, Force tolerance = 1.0eV/Å
#### Thermal Equilibration under NPT (40 ps)
velocity matrix create 300.0 5812775 mom yes rot yes dist gaussian # Initialize at 300 K
fix 1 matrix npt temp 300.0 300.0 $(100.0*dt) iso 0.0 0.0 $(1000.0*dt) # NPT ensemble, 0 bar pressure
fix 5 matrix temp/rescale 10 300 300 1 0.5
timestep 0.0001 # Time step of 1 fs
thermo 100
dump 1 all custom 100 dump_n1.lammpstrj id type x y z
run 10000
unfix freezeCNT_coating
unfix 1
#### Uniaxial Tensile Deformation in z-direction (NVT Ensemble)
timestep 0.01
fix 2 matrix nvt temp 300.0 300.0 $(100.0*dt) # NVT ensemble at 300 K
fix 3 all deform 1 z erate 1.0e-4 remap x
#### Output strain and stress info to file
#### For units metal, pressure is in [bars] = 100 [kPa] = 1/10000 [GPa]
#### p2, p3, p4 are in GPa
variable tmp equal "lx"
variable L0 equal {tmp}
print "Initial Length, L0: ${L0}"
variable strain equal "(lz - v_L0)/v_L0"
variable p1 equal "v_strain"
variable p2 equal "-pxx/10000"
variable p3 equal "-pyy/10000"
variable p4 equal "-pzz/10000"
fix def1 all print 100 "{p1} {p2} {p3} {p4}" file alloy.def1.txt screen no
dump 2 all atom 50 dump.final
thermo 100
thermo_style custom step v_strain temp v_p2 v_p3 v_p4 ke pe press
run 200000 ```