Hello all Lammps user,
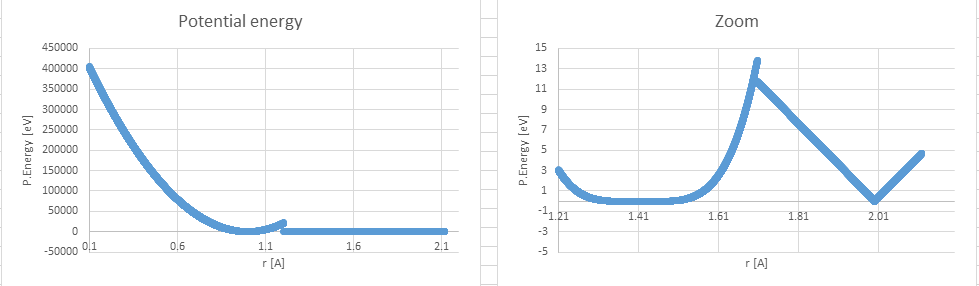
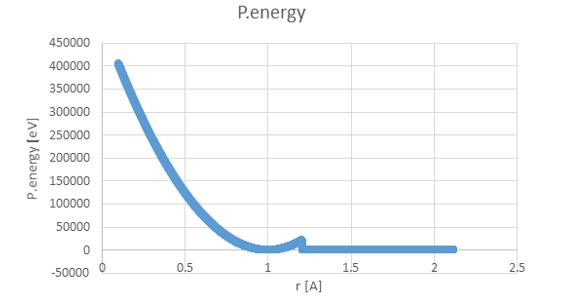
Im trying to set the potential as described in the attached picture (pEnergyWanted). It should be noted that the potential energy and the forces are not continuous and their values are huge up to r=1.21.
I wrote a matlab code which output file starts with:
Hello all Lammps user,
Im trying to set the potential as described in the attached picture
(pEnergyWanted). It should be noted that the potential energy and the forces
are not continuous and their values are huge up to r=1.21.
I wrote a matlab code which output file starts with:
~~~
# Exp potential
POT_LG
N 30000 RSQ 0.10011071 2.11803399
1 0.10011071 404900.36651812 899889.28932188
2 0.10022142 404800.74529309 899778.57864376
3 0.10033213 404701.13632492 899667.86796564
.
.
~~~
and in the code it is written:
~~~
pair_style table linear 30000
pair_coeff * * potential_D1000000_g40.txt POT_LG 2.1
mass * 1.0
pair_write 1 1 30000 rsq 0.10011071 2.11803399 potAfterLinearRSQ.txt POT_LG
~~~
I followed the guidelines in the manual for using pair_style table command,
i,e, I used many valued in table, used RSQ to avoid additional
interpolation, set linear style, and used relatively high inner cutoff (I
also tried to start from 0.5, same happened).
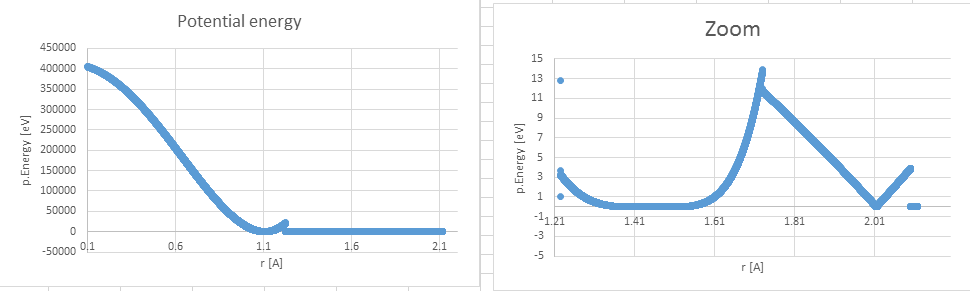
However, pair_write's outputs shows unwanted differences after Lammps
interpolation process (pEnergyOutput), which cause to error: "Pair distance<
table inner cutoff".
In addition I tried to set the inner cutoff to a value very close to 0 and a
new error appeared "Lost Atoms".
I guess it is because of the very inaccurate interpolation. How can I fix
this problem??
no. the problem is that your table is inconsistent. you state to have
an RSQ style table with squared distances, but the table is
equidistant, i.e. R style.
the r values in the table are ignored, instead they are generated from
the line at the top.
axel.
Axel, I tried to apply what you said (I change to R) as described below.
It seems to give better results (picture PEwithR) after r=1.21, but it is shifted and the energies are now even worse below that distance.
I tried to use 300,000 lines in the table (instead of 30,000), using linear and spline, and still I got the same results.
I also tried to use 1,000 (or 5,000) values up to r=1.21 and 20,000 values after that with R and RSQ, but it makes it even worse.
What do I miss?
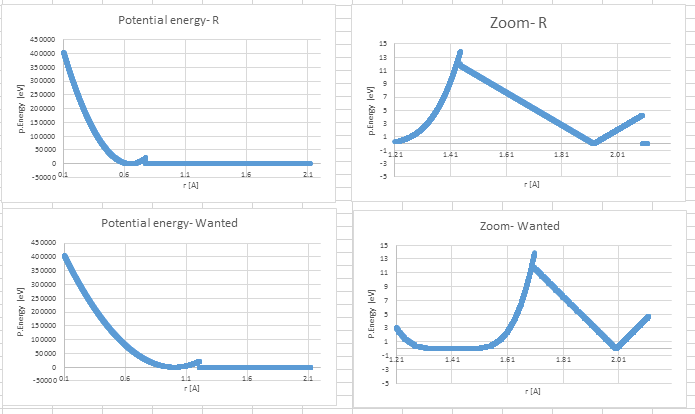
Axel, I tried to apply what you said (I change to R) as described below.
It seems to give better results (picture PEwithR) after r=1.21, but it is
shifted and the energies are now even worse below that distance.
I tried to use 300,000 lines in the table (instead of 30,000), using linear
and spline, and still I got the same results.
I also tried to use 1,000 (or 5,000) values up to r=1.21 and 20,000 values
after that with R and RSQ, but it makes it even worse.
What do I miss?
i don't know. i am not clairvoyant. my guess is that your potential
table is still inconsistent.
axel.
I’ll just add that you willl not be able to do consistent dynamics
(e.g. conserve energy) if the E and forces are non-continuous.
Steve
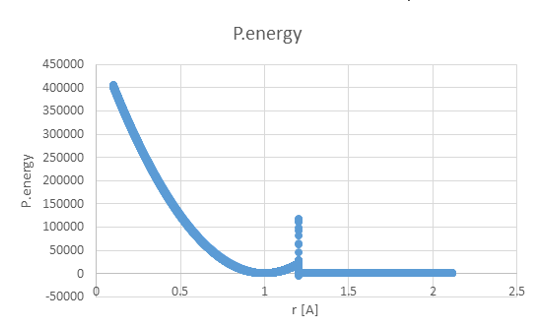
Thanks for the replies, it helps a lot!
I changed the potential file to:
The pair distance error means that 2 atoms are closer than the
minimum distance in your table. Hence LAMMPS does
not know how to compute the energy/force.
When LAMMPS creates the interpolated table it has no
choice but to bridge any discontinuities with energies/forces
inbetween those value (e.g. steep changes).
Steve