Hello everyone
i am currently identifying a weird thing for the results dumped by
compute 6 all stress/atom NULL
i used a simple input script with pair_style granular to study the results for stress tensor of each atom. to be simple, only perfectly spherical grain is used in this case.
but it seems that i got clearly weird results.
my pair_style pranular is
pair_style granular
pair_coeff 2 2 hertz/material 7.0e10 0.1 0.2 tangential mindlin_rescale/force NULL 1.0 0.1 rolling sds 10000 5000 0.3
pair_coeff 1 1 hertz/material 7e6 0.5 0.5 tangential mindlin_rescale/force NULL 1.0 0.5 rolling sds 0 0 0
pair_coeff 1 2 hertz/material 1.679785e7 0.223607 0.316228 tangential mindlin_rescale/force NULL 1.0 0.223607 rolling sds 0 0 0
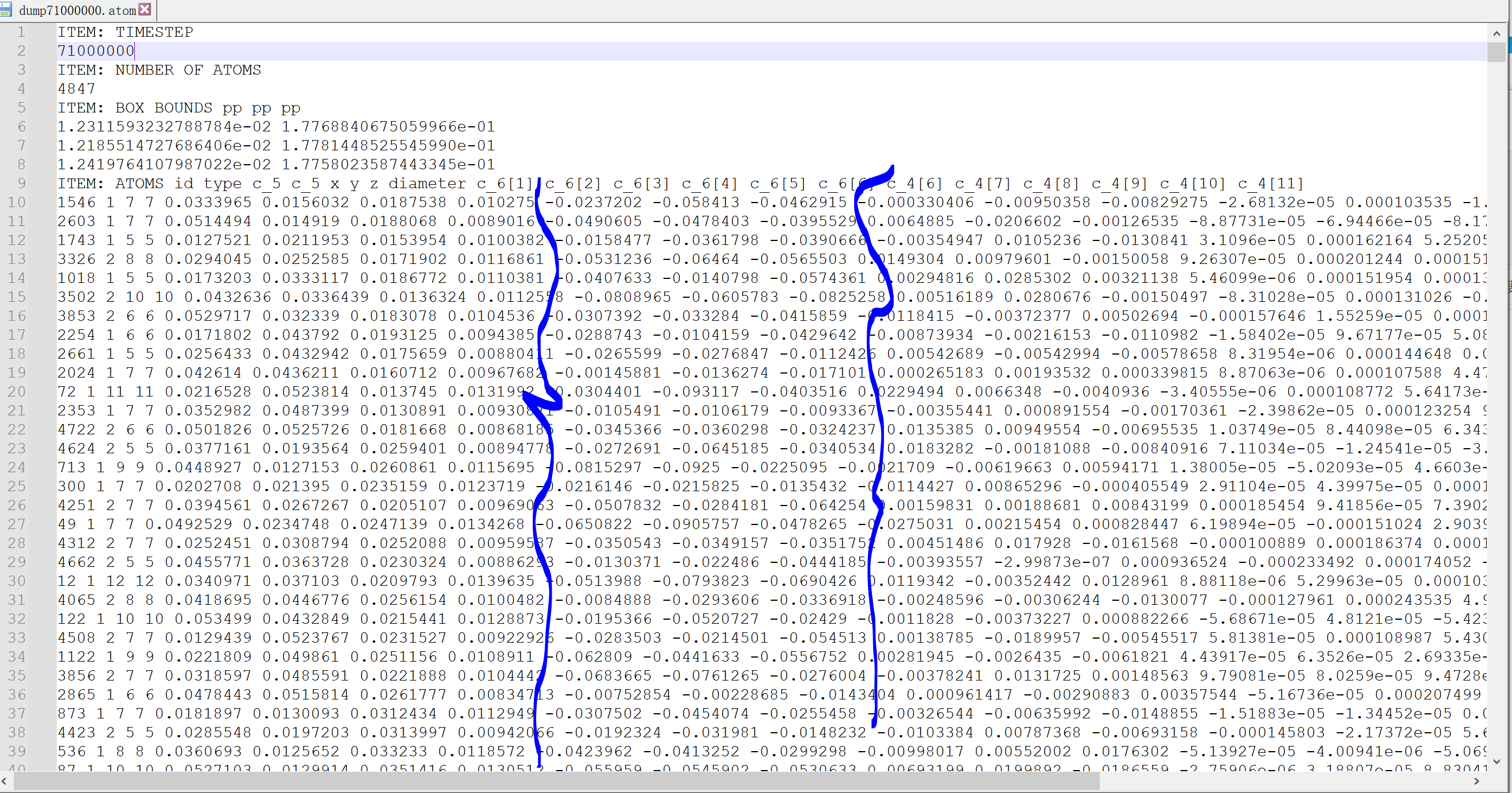
then i used this dump custom to got the stress tensor for each atom
dump 2 all custom ${interval_dump} check_atom/dump*.atom id type c_5 c_5 x y z diameter c_6[1] c_6[2] c_6[3] c_6[4] c_6[5] c_6[6] c_4[6] c_4[7] c_4[8] c_4[9] c_4[10] c_4[11]
therefore the c_6[1] c_6[2] c_6[3] c_6[4] c_6[5] c_6[6] should be my stress tensor for each atom. but
i got very small and negative results, which may not really right, especially for the stress tensor xx, yy, zz. i attched the screenshot for my dump files. the stress tensor xx,yy,zz are negative and very small. to my own understanding. it should be not right. is this maybe a potentially bug? at least the stress tensor xx, yy,zz should be positive ?
hope you have a nice day
my apologies, i should highlight that i used June 15 develop version lammps
You are thinking the stress should be positive as in tensile? There is an inverted sign between compute stress/atom and compute pressure.
Hi jtclemm
many thanks for your response and time. it is really helpful.
i am thinking the stress of atom should be positive as in compression. and the magnitute of these stress tensor is too small, which are just around 0.02 in xx yy zz direction, something like this. is this magnitute make sense?
i used to look the stress tensor of atom in a old version of lammps, i always got stress tensor around 100, which is also positive in compression. it seems a big gap,
would you mind share any idea for that?
hope you have a nice day
best regards
Are you just missing a sign convention? e.g. I think a non-zero (positive) kinetic energy will lead to a negative stress for a system under compression compute stress/atom command — LAMMPS documentation
Otherwise I might not understand your question.
Hi @Tom_lammps,
Does it seems that you got weird results or did you get clearly weird results? We do not know about your system. Why did you expect “positive values as in compression”? Why is a value of 0.02 too small? What is your system? You say you get a value of 100 (which units?) in older version. Was it on the same system? What else could make a difference (units? bugs? pair coeff?)?
Also remember that according to the manual:
It is also really a stress*volume formulation, meaning the computed quantity is in units of pressure*volume. It would need to be divided by a per-atom volume to have units of stress (pressure), but an individual atom’s volume is not well defined or easy to compute in a deformed solid or a liquid. See the compute voronoi/atom command for one possible way to estimate a per-atom volume.
So these are stress tensor contributions, but not stress values per-se.
Hi jtclemm
many thanks for your response and time, its really helpful.
i can see the reason, the old verision of lammps used a different unit for the output of the stress tensor.
my apologies for any caused troubles and inconvenicens.
hope you have a nice day
Hi Germain
many thanks for your help and response, i can now see the reason, the unit used in my old version of lammps is stress value per-se. while now its stress tensor contributions.
many thanks for your time, my apologies for any caused inconveniences.
hope you have a nice day
best regards