I am trying to create a Pourbaix Diagram for the Nb-C-O system. My goal is to add a new solid (Nb2CO2) to the Pourbaix Diagram whose formation energy I have calculated from VASP (-33.33 eV). I tried using using the PDEntry approach. However, I encountered an error “‘PDEntry’ object has no attribute ‘phase_type’.” Seeking help to resolve this issue and successfully add the new solid to the Pourbaix Diagram.
/usr/local/lib/python3.8/dist-packages/pymatgen/analysis/pourbaix_diagram.py in init(self, entries, comp_dict, conc_dict, filter_solids, nproc)
512 self.pourbaix_elements = self.pbx_elts
513
→ 514 solid_entries = [entry for entry in entries if entry.phase_type == “Solid”]
515 ion_entries = [entry for entry in entries if entry.phase_type == “Ion”]
516
/usr/local/lib/python3.8/dist-packages/pymatgen/analysis/pourbaix_diagram.py in (.0)
512 self.pourbaix_elements = self.pbx_elts
513
→ 514 solid_entries = [entry for entry in entries if entry.phase_type == “Solid”]
515 ion_entries = [entry for entry in entries if entry.phase_type == “Ion”]
516
AttributeError: ‘PDEntry’ object has no attribute ‘phase_type’
@rkingsbury Thank you for your prompt response. The tutorial notebook you provided was indeed helpful, and I’ve successfully developed the code to append Nb2CO2, which is attached here. However, I have two questions that I would appreciate your guidance on:
Since I calculated the formation energy from DFT, I suspect I may need to apply some corrections to align the formation energy with the existing entries from the Materials Project (MP). Could you please advise me on how to proceed with this correction?
I noticed that certain entities, such as ions, related to the appended entry (Nb2CO2) are not present in the Pourbaix diagram. Specifically, I couldn’t find entries for Nb2O3- or Nb2(OH)5. How should I handle such entities when dealing with the Pourbaix diagram?
Thank you again for your assistance. I’m looking forward to your valuable input on these matters.
*# Get all pourbaix entries corresponding to chemical system.*
entries = mpr.get_pourbaix_entries(["Nb", "C", "O"])
*#Additional entries*
solid_ent_e= PDEntry(composition="Nb2CO2", energy=-33)
pb_solid_entry_e = PourbaixEntry(solid_ent_e)
entries.append(pb_solid_entry_e)
You are correct about needing to apply energy corrections. See MP docs. Practically speaking, once you have created ComputedEntry from your new DFT calculations, you then need to apply the MaterialsProjectAqueousCompatibility scheme.
from pymatgen.entries.compatibility import MaterialsProjectAqueousCompatibility
aqcompat = MaterialsProjectAqueousCompatibility()
aqcompat.process_entries(<your list of DFT ComputedEntry>)
The entries will be modified in-place with necessary energy corrections. Afterwards, you can inspect the energy_adjustments attribute of any of those entries, and you may see some corrections applied.
Adding ions is a bit more complicated, and unfortunately not very well documented. Basically, you need to first determine the free energy of those ions relative to some reference solid for which you have DFT energy and you can also find experimental energy. Then you position the ions relative to DFT energies on the Pourbaix diagram.
For example, suppose you go to literature and find that Nb2O3- ion has a free energy of -100 kJ/mol, and solid Nb2CO2 has a free energy of -150 kJ/mol. Note that both values are experimental. So experimental data says that the ion free energy is 50 kJ/mol higher than that of solid Nb2CO2
Next, find the (corrected) DFT free energy of solid Nb2CO2 on your PourbaixDiagram. Add 50 kJ/mol to it to determined the adjusted free energy to use for your ion in the Pourbaix Diagram.
Finally, create a PourbaixEntry for the new ion using that free energy, and include it in the list of entries when you build your Diagram.
I hope this makes some sense. You can study the code of the get_pourbaix_entries method to see this in action for the ions that we have in the MP database.
@rkingsbury Thank you for your help. I have made the necessary modifications to my code, but I encountered some errors. Please have a look at this error.
Also, can you please confirm whether in PDEntry, the energy represents the formation energy of the structure? Specifically, I am unsure if the energy value should be given as the formation energy per atom (e.g., -0.1 eV/atom) or the formation energy of the formula unit (e.g., 5 * -0.1 eV) for a material with a formula unit of “Nb2CO2”?
entry_elements = mpr.get_pourbaix_entries(["Nb","C"])
#Additional entries
solid_ent_e= PDEntry(composition="Nb2CO2", energy=-4)
with MPRester(MP_API_KEY) as m:
entry_O2 = m.get_entries('mp-723285')
entry_H2 = m.get_entries('mp-24504')
entry_H2O = m.get_entries('mp-697111')
# define list of entries
entries = [entry_elements, solid_ent_e, entry_O2, entry_H2, entry_H2O]
# Compute the 'AqueousCompatibility'
aqcompat = MaterialsProjectAqueousCompatibility()
entries = aqcompat.process_entries(entries)
# Construct the PourbaixDiagram object
pbx = PourbaixDiagram(entries)
plotter = PourbaixPlotter(pbx)
plotter.get_pourbaix_plot().show()
HI @nihal , sorry for the delay in my response here. I suspect the reason for your error is that the entries list you built contains both PDEntry and PourbaixEntry (which are returned by get_pourbaix_entries. When applying a compatibility scheme, you want entries to comprise only ComputedEntry. A ComputedEntry is quite similar to a PDEntry, except it carries additional data related to how the calculation was done and provides facilities for energy corrections.
In your example, m.get_entries should always return ComputedEntry. So I would replace m.get_pourbaix_entries(["Nb","C"]) with m.get_entries(["Nb","C"]).
As to your question - my understanding is that a PDEntry can be populated with either free energy of formation OR electronic energy (DFT energy), but all the PDEntry used to make a phase diagram must use the same one. ComputedEntry, on the other hand, are intended to hold electronic energies.
Once you apply the corrections, you’ll then need to follow some additional steps to get to a Pourbaix diagram. In brief, you use the corrected energy to make a Phase Diagram and compute the formation energies of each material, then you use those formation energies to create PourbaixEntry.
See the steps in the “Building a Pourbaix Diagram Manually” section of this tutorial notebook (which I think you have already seen?) for more detail.
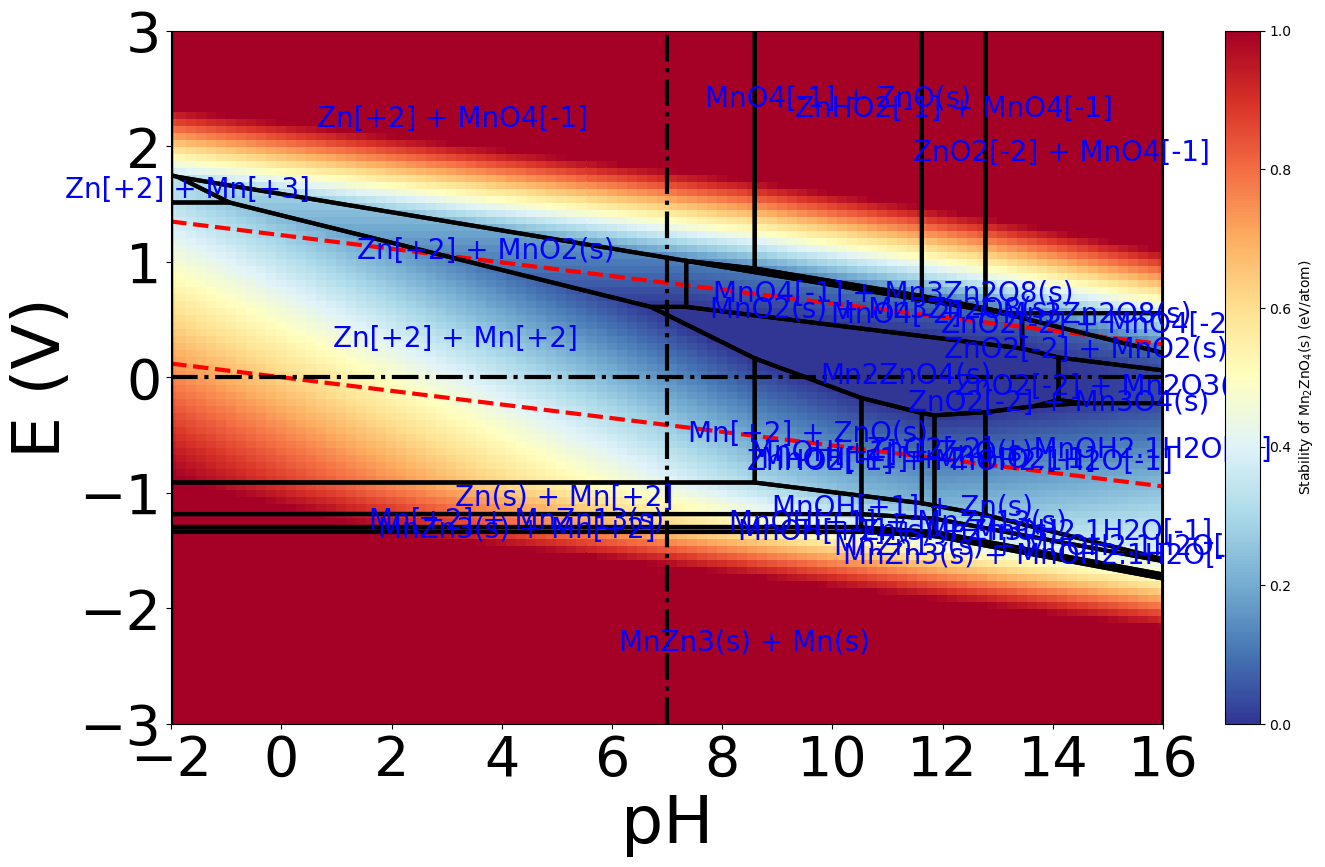
When I see this plot, Mn2ZnO4(mp-18751-GGA+U) show ΔGpbx < 0.5 eV/atom meaning it aqueous stability, but I can not see the exactly value of ΔGpbx
How I can print out the value of ΔGpbx for Mn2ZnO4(mp-18751-GGA+U) using pymatgen code?
Thank you so much for helping!
Dear sir,
I have a question about this, I was trying to following the tutorial you mentioned. However, it goes run if I use mpr.get_pourbaix_entries. The code is
from pymatgen.analysis.pourbaix_diagram import PourbaixDiagram, PourbaixPlotter
from mp_api.client import MPRester
Please run pip install --upgrade mp-api mpcontribs-client to make sure you are using the latest versions. I don’t know the exact cause but I suspect something changed in the packages and your versions are no longer compatible.
Much thanks to your reply. Here is another question I want to consult. I want to take a large amount of Pourbaix simulation by the mpr.get_pourbaix_entries(). It seems to require large and long-running queries. And I also notice that I can make a local copy of the ion database in the page of API. However, when I check the Endpoint of data document, I didn’t find the property related to the ion data.
Besides, I also check the source code of the “get_pourbaix_entries” it shows that
url = “/pourbaix_diagram/reference_data/” + “-”.join(chemsys) ion_data = self._make_request(url) ion_ref_comps = [Composition(d[“Reference Solid”]) for d in ion_data] ion_ref_elts = list(itertools.chain.from_iterable(i.elements for i in ion_ref_comps))
Even if I make the local copy of the ion database, when I try to use the get_pourbaix_entries(). It requires to make the query to the online database.