Short problem description:
I am trying to equilibrate the pressure to 1 atm in the direction perpendicular to the electrodes in an electrochemical cell, however I don’t understand how to interpret the much lower pressure that LAMMPS reports with the thermo output. This is basically a thermodynamic question, but goes into how LAMMPS calculates the necessary quantities to calculate the pressure when I constrain movement, bonds and angles of atoms and molecules.
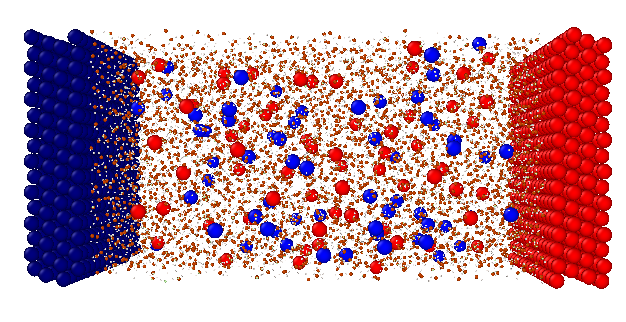
System
An example of the system can be seen in the picture below. I define two electrodes with electrolyte in between. I use a rigid TIP4P water model, but add the fourth M-sites explicitly, (so that it works together with the ELECTRODE package which I use after equilibration).
I constrain the bonds and angles of the TIP4P molecules by using the fix rigid/nvt/small molecule and I freeze the electrodes with the following two commands:
fix … npt temp 298.15 298.15 100 z 1 1 1000 couple none dilate all.
From visualizing the trajectory and the bulk density of the electrolyte everything seems fine. The system seems to equilibrate and fluctuates around a pressure of roughly -2000 atm. This is not the 1 atm I asked for, however I didn’t ask it to output the pressure in the z-direction either.
I am wondering however if the pressure calculation can be correct, considering the fact that the pressure is calculated as (P=pressure, E=total energy, V=volume):
P = - (dE/dV)
The number of atoms is also part of the calculation (not shown in this equation).This is dubious in my case, as I am using a four-point water model thus I have more particles in my system than when using a ‘normal’ 3-point water model. Further, by freezing the electrodes and using a rigid water model I also affect the total energy of my system considerably.
I have two questions:
What would be the best way to output the pressure in the z-dimension?
I tried outputting Pzz, but from my initial tests, Pzz seems to fluctuate around -1200atm after 50ps. Also, I did see an example in the documentation for a box with dimensions that do not change during the simulation by summing the stress per atom in the z direction in bins, but this only works for fixed boundaries.
With all of the assumptions above, when I say: ‘LAMMPS simulate a pressure of 1 atm in the z-direction’, can LAMMPS accurately simulate 1 atm of pressure?
What I mean by accurate is, would it correspond to the experimental situation in which I would not have frozen electrodes in my system? I could try using the temperature of the electrolyte for the pressure calculation which prints the pressure to my logfile, but that wouldn’t change the barostatting of the Nose-Hoover thermostat itself. I cannot exclude the electrodes from being integrated, because then the system cannot expand in the z-direction.
The default pressure compute that is used for the thermodynamic output is this one: compute pressure command — LAMMPS documentation
As you can see the pressure is computed from virial stress which can be derived from the forces when those are accumulated. This this is computing a stress * volume term, you need to divide by the volume. The global pressure compute thus uses the virial contributions from all atoms and divides by the entire volume. For your needs, it would be more consistent, to only look at the contributions from the mobile atoms and for the volume those atoms are in. … and this is where the problems start: the total box volume is an input parameter and thus well defined, the volume of a group of atoms, less so, thus it is not so straightforward how to determine this. Since you have a dense system you could use Voronoi tesselation. Or you could run a bulk simulation of your mobile material without the walls and determine its density from a bulk system and from that the average volume per atom (or molecule). How to compute a pressure from the per-atom stress for all or a subset of atoms is outlined in the docs for the compute stress/atom command — LAMMPS documentation
The behavior of systems on the atomic scale can be quite different from macroscopic experiments since your time and length scales are very different. The ratio of bulk versus interface is very different and thus it is often more productive to just simulate the bulk, unless you are specifically interested in the interface and then you could just simulate a sufficiently thick film absorbed on one of the electrodes and then the force field will work out the proper density by itself.
That is about as much as I can provide, since this is not my area of research. Perhaps @srtee, who is one of the developers of the ELECTRODE package, has some more specific insight. For that, it may be helpful, if you could explain in more detail what you want to learn from the simulation(s) once you do have a properly equilibrated system.
This is how I’ve always done it. When equilibrating my electrode systems, I usually add an “accordioning”* step where I move the electrodes apart in z and back together (without constant potential) and check that the “bulk” density (usually the middle 20% of the box, which should be far enough from the electrodes that the interfacial density fluctuations are damped, and this can be confirmed via the usual fix ave/chunk) is equivalent to a fully periodic NPT simulation at the desired temperature**.
Once I’ve decided on a suitable inter-electrode distance, one of the quantities I monitor during equilibration (with fixed electrodes and constant potential) is this “bulk” density, which usually is not affected by the constant potential (which makes sense, since the entire conceptual assumption is that the “bulk” is not too affected by the non-bulk-like interfacial dynamics).
This procedure is described in my papers. I can’t remember if I included an equilibration script in the Zenodo accompanying my paper from April this year – shame on me if I didn’t and I’ll have to rectify that, and happy to attach the script here if so.
I’ve never been hassled by reviewers on this point (maybe I will after this post!), but if I were, my rationale would be that the whole point of an MD simulation is to obtain information I can compare (even qualitatively) to experimental systems, and if a force field (1) replicates bulk densities in a fully periodic NPT simulation (2) has the same bulk density in a large enough volume between electrodes then that’s enough evidence that it does model electrolyte at a suitable pressure.
I freely admit that this procedure would fail if someone wanted to simulate electrodes close enough that nanoconfinement affects the whole system and a “bulk-like” region no longer exists. My first instinct would be to compute group/group the interactions between the electrolyte and electrode and fall back on the physical definition of pressure as average force per area.
*I’ve never called it that, but obviously I’ll never call it anything else from now on.
**I always do a full periodic NPT simulation anyway, to make sure the force field is behaving correctly by checking RDFs and diffusivities.
First of all, I want to thank the both of you for taking the time to answer my questions. I really appreciate your help!
The idea of first equilibrating the mobile part of my system and checking for the bulk density sounds reasonable to me. Also, Shern, I loved the term you came up with: accordioning your system.
However, in the files you shared on Zenodo I could not find an equilibration procedure. In an older paper I did find the following explanation:
What I understand from this is that you equilibrate each simulation box ‘by hand’ for a specific concentration. I am not sure if you have written it in such a way that LAMMPS itself stops if the right bulk density has been found (I am not even sure whether LAMMPS could do that or not), but this sounds inconvenient to me, as I am planning on running hundreds of simulations.
Based on both of your answers, I think I will skip the npt equilibration, as I cannot read out the pressure in any sensible/straightforward way and I already account for the right bulk density anyway by adding:
extra water molecules to account for the higher water density near the interface, and
shielding ions to account for the charges that I put on the electrodes later
to my initial simulation boxes. I checked the resulting bulk densities when running the CONQ method for different concentrations and they seem to relate more or less to the experimental densities for aqueous solutions of alkali chlorides. Also, for my research I am interested in qualitative relationships so if the resulting bulk concentration doesn’t perfectly match the concentration I was aiming for, I simply report on the resulting concentration instead.
Thank you again for your help and have a great day.
P.S.: Axel, thanks for the suggestion to use Voronoi Tesselation to calculate the effective volume of an atom/molecule in my simulation box so that I can get the pressure in my system. I found some beautiful pictures of Chinese vases! But all jokes aside, I think you meant something to use like this paper: https://www.sciencedirect.com/science/article/abs/pii/S0167732221028981. It sounds like an elegant and creative solution to the problem, but maybe also a bit overkill for what I am trying to accomplish.