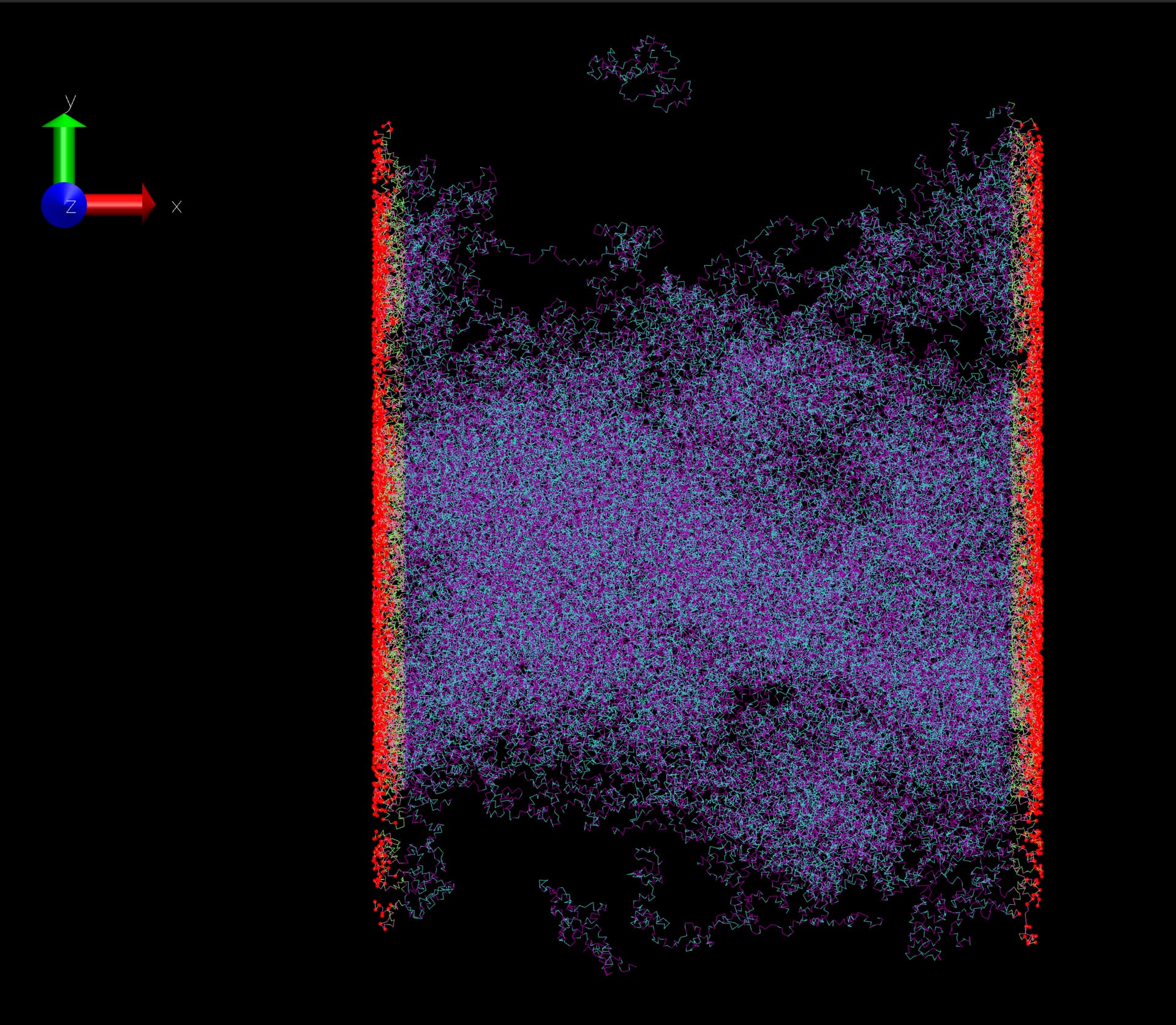
I have a polymer system to simulate using a coarse-grained model, which includes tabulated non-bonded, bond, and angle potentials. The boundary condition is periodic in the Y and Z directions but non-periodic in the X direction (m p p). To create the non-periodic boundary condition in the X direction, we introduce auxiliary points (colored in red), which are connected to the polymer chains using a harmonic bond potential. During the simulation, these points are fixed in space. I am trying to maintain the pressure of the box at 1 atm in the Y and Z directions. To do this, I can use the barostat in LAMMPS, such as fix 1 all press/berendsen y 1.0 1.0 z 1.0 1.0 1000.0 or the npt command.
The problem is that I need the polymer system to be at 1 atm excluding the red points, but when the fix press/berendsen runs, it likely includes the virial contribution from the stress of the fixed points (red points). Am I correct? If so, how can I resolve this issue?
I think LAMMPS always includes the forces experienced by every single structural unit (atoms or superatoms) of the system in the virial term. There are ways you can adjust the temperature-dependent term to concern only one group of structural units, but not for the virial, if I remember correctly.
There has been some discussions in the forum about it not being correct to consider only a specific portion of the system when computing the pressure, so even if you manage to make the pressure calculation (including the virial term contribution) concern only a group of atoms, it might not be meaningful. I dont know much about that to come up with a beautiful reasoning or insight on your case.
If you want to proceed and converge to a thermodynamic state in which only the pressure stemming from the polymer superatoms corresponds to 1 atm, one way that comes into my mind (although maybe not the most efficient one), would be to create an external code to compute the values of Pxx, Pyy and Pzz only taking into consideration the polymer superatoms and adjust correctly the target pressure in the barostat to a value that ultimately yields a thermodynamic state in which “the pressure of the polymer superatoms” corresponds to 1 atm. But then again, you would need to see if this “partial pressure” notion would correspond to something meaningful in your case.
To get input data for that code that you would have to build, you could save a force-containing trajectory. Ofc you would need to later exclude the contribution coming from the interactions with the fixed atoms. You can do that by doing a rerun of that trajectory afetwards and saving a new one or dedicate a part in your code for doing that exclusion.
I think (kind of out of my head) that one of the problems of considering only part of your system when computing the pressure is the fact that in the “real system”, where all the atoms exist, the actual pressure corresponding to that thermodynamic state is not “partial one” you are considering. So you would kind of be trying to say your results corresponds to a thermodynamic state of pressure X when the real pressure the results stem from is one in which the polymer is at a different pressure.
Maaaaybe if you have a lot of “bulk” superatoms (i.e. ones that are not interacting with the walls), your value of “partial pressure” would be more consistent with the reality of your system.
Another thing to consider is that the computing of pressure seem to involve the ghost superatoms. So I dont know if Pxx for example would be meaningful since there is no periodic boundaries.
This is not a good idea. If your “red atoms” are immobile, then you should not us fix npt. Either those positions are rescaled by fix npt which results in unphysical geometries by squeezing or stretching them beyond they “normal” geometry. And if you exclude them from rescaling you will have overlapping atoms at the box boundaries.
The only degree of freedom to adjust the pressure in the system that you should be using is along the x axis. You can make those atoms immobile in y and z with fix setforce NULL 0 0, but keep them mobile in x using fix aveforce 0 NULL NULL. You can use a value different from 0 in order to add force to the “red atoms” so they either squeeze or expand the system in x. Given that you know the geometry of your system and you are not changing y and z dimensions, you can compute the force per area and thus the average force per atom that you need to add. Since you are effectively ignoring the forces between the “red atoms”, you can us neigh_modify exclude to remove the force computation (and thus contribution to the stress tensor) between those atoms, but keep the interaction with all other atoms.
If you want to go even more fancy, you can have a look at fix controller which could be used to adjust the value of the added force until a target pressure is achieved. This command is quite powerful but requires careful testing and tinkering with the setting to achieve the desired results. For example, you are not limited to the global pressure, but can also adjust to the average pressure in y and z and so on.
I was not the primary investigator in this work, but we faced a similar issue. If I remember correctly, the procedure for system creation is outlined in the paper.
I am using stress/atom to compute the pressure instead of reporting Pxx, Pyy, and Pzz, so I do not think I should be worried about the ghost atoms. The problem with “fix deform/pressure” is that the harmonic bond interaction between the fixed atoms (red points) and the polymer chain itself is included in the virial part. As you suggested, one way would be to set the different pressures for the whole system in a way that the motion particles get the real 1 atm. However, I am looking for a better solution since I will have many different systems of varying sizes.
I plan to do a uniaxial tensile test by moving the red-fixed particles in the X direction while keeping the Y and Z directions at 1 atm. The red-fixed particles do not interact with each other, and there is no pairwise interaction between them and the polymer system (pair style zero for red particles and polymer). Each red particle is only connected to a single polymer chain using a harmonic bond.
The command “fix 2 all deform/pressure 1000 y pressure 1.0 0.001 z pressure 1.0 0.001 normalize/pressure no max/rate 0.0001” works for the system by rescaling all particles, including the fixed particles, in a periodic direction. I calculated the stress for the mobile particles in the Y and Z directions, which is around -11 atm instead of 1 atm. I think this means that the harmonic bond between the polymer system and the red-fixed particles is included in the virial part.
If I use the neigh_modify exclude command, will this also exclude the force contribution of the harmonic bond on the stress tensor? If not, what would you suggest I do? Is there a correct way to do this? Should I change the source code to exclude the harmonic bond from the pressure calculation and then use the “fix deform/pressure” command? I guess when the “fix deform/pressure” command runs, it calculates the pressure for the whole system in the Y and Z directions and then tries to rescale the box.
I didn’t know about the application you aim for your system and posted this article because the problem in setting the system was the same. The interesting part of the article was in the methodological aspect (II.D and E, as well as III.A).
I couldn’t type on a proper keyboard so I might have been a bit short in my answer, but the way we found an suitable system size along the grafting direction was by comparing with equilibrium properties of bulk polymer, since defining local pressure and equilibrating it in anisotropic non-periodic system can be tricky in practice (see @akohlmey’s answer).
This system has mixed periodicities and is nanoscale and (appears) highly inhomogeneous, all of which complicate the interpretation of “pressure” from instantaneous configurations.
If I were in your situation, I would give up on any kind of dynamic pressure control for the time being. Use fix move to move those red “anchors” apart, and then back towards each other, at prescribed velocities, and obtain the stress as a function of strain. You can much more easily analyse potential sources of error:
Isolate any hysteresis by equilibrating the system for a while in maximally stretched / “squashed” states, and then monitoring the forwards-backwards curve differences, either confirming the absence of hysteresis or its magnitude
Determine rate dependence by conducting simulations at varying stretch / “squash” rates.
How should I deal with the system in the Y and Z directions? During the uniaxial tensile test in the X direction, the pressure of the mobile atoms (excluding red points) in the other two directions has to be at 1 atm. Do you think it is practical to remove the contribution of the red points from the virial part of the stress tensor and then leave the box deformation in the Y and Z directions to the NPT ensemble (with the modified global pressure in the Y and Z directions)?