Lammps version: LAMMPS (29 Aug 2024).
Platform: Computing cluster
Dear users,
I am trying to reduce the number of particles needed to simulate the recombination of radiation defects in a diamond. Since this is a simulation of a crystal the defected structure only contains a small number of relevant particles (aka the point defects).
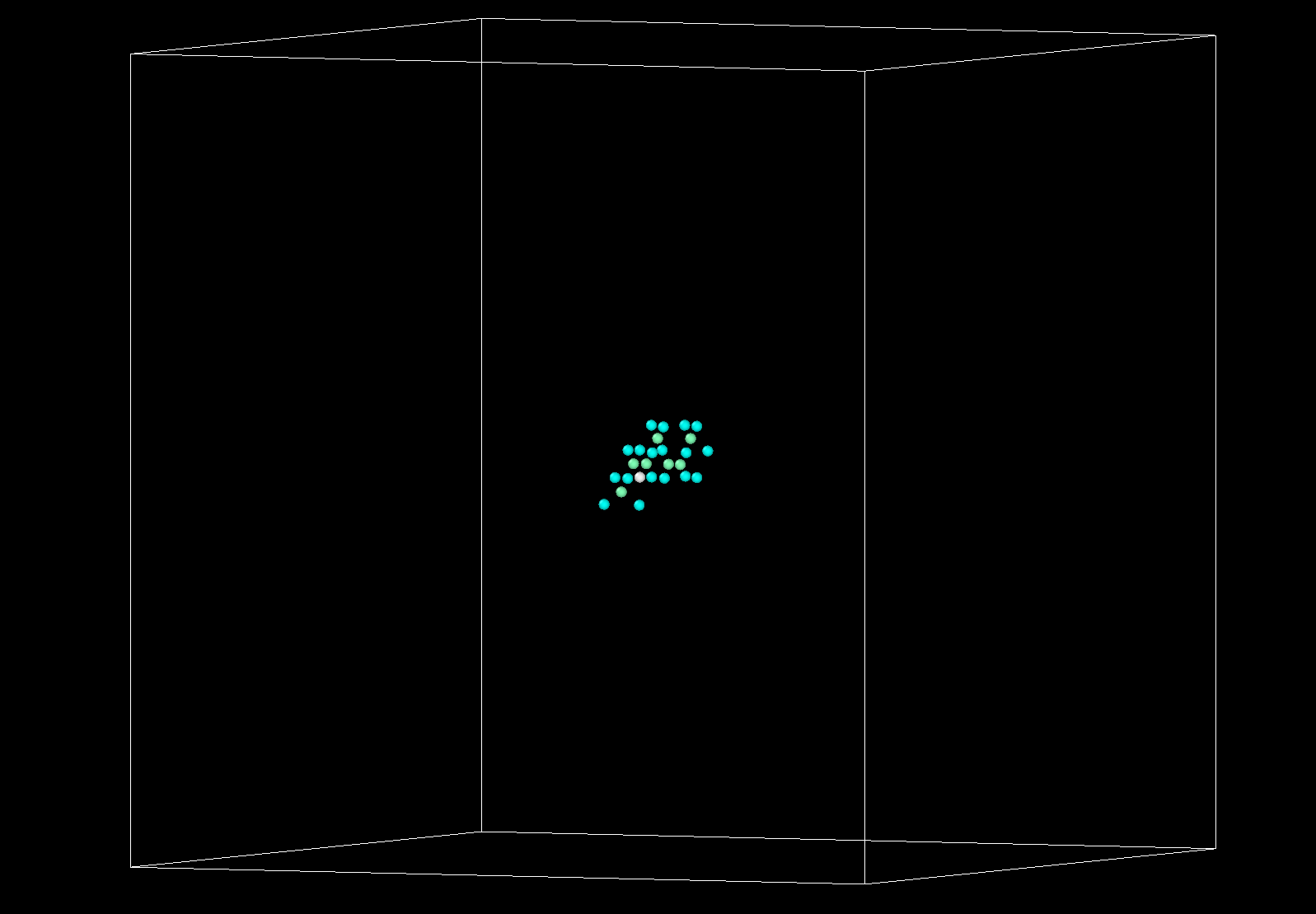
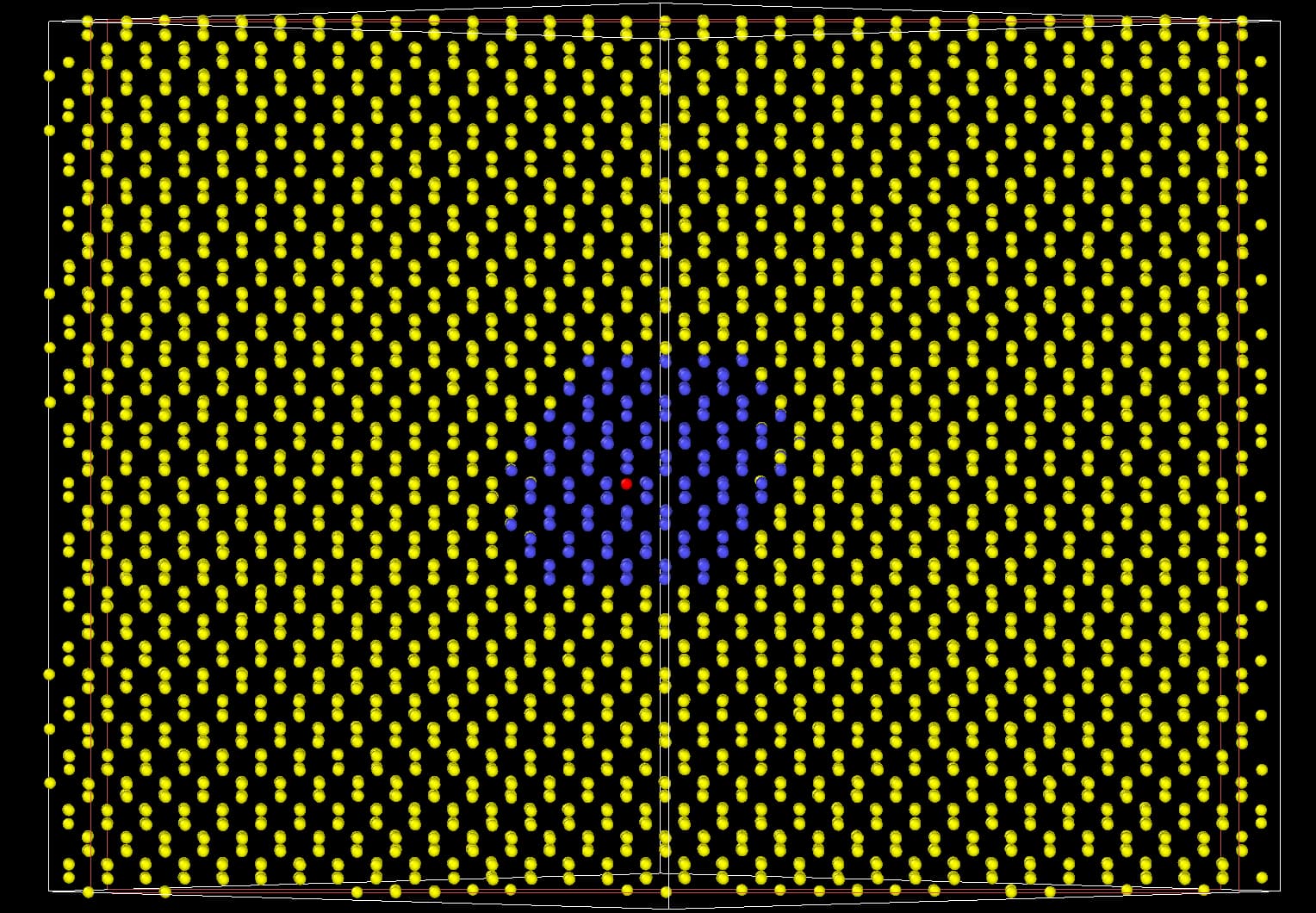
I therefore excluded from the neighbor lists all the particles distant more than 2 nm appart from the defect. I left them as ghost particles for the included particles. The subset of mobile particles can be seen here:
Additionally I constrain the movement of the ghost particles to an harmonic potential using the fix tether. I applied a fix langevin + fix thermostat to all particles
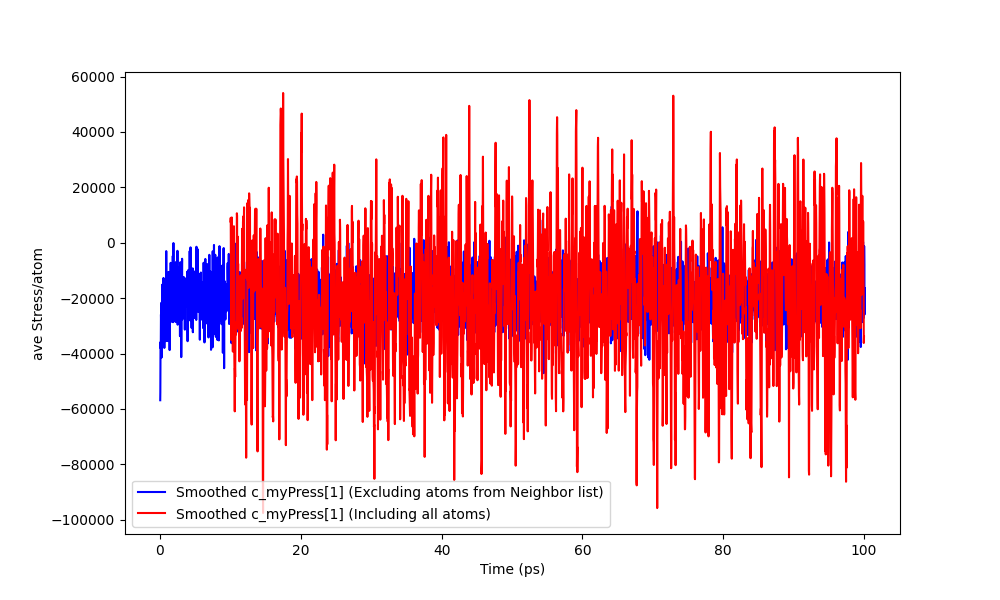
I computed the thermodynamic output using the atoms included in the neighbor lists only. I was able to obtain accurate temperature fluctuations (the time series has the same frequency, average value and std). However for the average stress on the mobile atoms the time series does not have the same standard deviation nor frequency as it can be seen in the following figure:
I have two questions: Is it hopeless to control the stress over a subset of mobile atoms having such constrained boundary atoms ? Is there any fix that could control the average strain of a subset of atoms?
This doesn’t make sense since fix langevin already acts as a thermostat.
1 the stress comes from the forces which you have removed. So I don’t see a clean way to get those back without restoring the interactions.
2 you would have to come up with thorough derivation of how to determine this.
I would suggest two different approaches: 1. getting access to more CPU power these days is easier than ever in my (30+ years of) experience and LAMMPS does have good strong scaling and good support for GPU acceleration. 2. I would suggest looking at r-RESPA as an option to save time. Some time ago we added a feature to allow less frequent force evaluation for coarse grain to coarse grain interactions for simulations that embed all-atom systems in coarse grain particles. You could do something similar by evaluating the forces of the embedding particles beyond a certain cutoff less frequently. That should give you some savings as well and on a more proven theoretical foundation. My main concern would be that this would suppress some phonons. But then again your proposed approach is far more drastic. In combination with my first suggestion and carefully chosen load balancing this has the potential of a significant speedup.
That cluster of particles is almost certainly small enough that neighbouring atoms’ instantaneous stresses are significantly correlated. This means the “average per-atom stress” is really an average over a very small sample size of “independent” stresses – of which you are further taking a subset.
It is not surprising for the average of very few independent variable copies to have a significantly larger standard deviation – that’s the Central Limit Theorem! By contrast, particles’ instantaneous kinetic energies are much less correlated (especially with a Langevin thermostat which explicitly decorrelates all particles via thermal noise), so the variance in temperature is genuinely thermodynamic and less affected by system size.
To my eyes the blue and red graphs have statistically similar averages. And you can’t take each separate snapshot in time as an uncorrelated sample either – you must use block averaging for a halfway decent estimate of standard error.
As for pressurising a subsystem – you MAY be able to accomplish this by simulating a bulk, non-defective system at your desired pressure, obtaining the reduced (shortened) lattice constant at that pressure, and then modelling your “defect environment” as particles harmonically tethered to a lattice with said pressurised lattice constant (an “Einstein crystal”, because we clearly didn’t name enough things after that guy).
You can then make a reasonable case that the funky particle cluster in the middle of the Einstein crystal background is in fact held, on average, at the desired pressure, especially if (1) the time-averaged displacement and stress per-atom remains homogeneous throughout the crystal background (2) your results match up with a few expensive fully-pressurised runs.
As far as I can tell, you will also have to tune the spring constant of the Einstein crystal to achieve the correct pressure at your desired temperature, but that can be done relatively simply (by measuring the pressure as a function of spring constant at your specified lattice spacing and temperature).
Yes. Sorry for that. I meant to say fix langevin (thermostat) + fix nve (integrator).
Indeed, this is only part of a bigger project involving larger simulations. By selecting which atoms we simulate in this equilibrium system we hope to acces larger simulation scales with a given number of processors. But indeed, if we cannot fix this stress issue we might just go with the usual pbc large box.
Thank you for this suggestion. I will take a look at it.
Indeed. My approach is more similar to what @srtee suggested. If the phonons are a problem I am wondering if my approach makes any sense at all, even if I chose to include more atoms. Or is it only when I apply periodic boundaries that I will obtain the right phonon spectra? Many of the calculations that I have read about, specially involving expensive techniques such as DFT, use small boxes. Following your argumentation I would imagine that the stress generated by e.g a defect would overlap with themselves.
This sentence has me a bit confused, because you comment that the stresses are “correlated” and independent. The value that I am monitoring is the stress/atom averaged over all the “mobile” particles at every thermo output step.
Both blue and red curves where computed over the same set of atoms. The difference is that, for the red curve the embedding atoms where time integrated while for the blue curve they where tethered. The sample size is the same.
Indeed the temperature timeseries obtained in the same fashion was much more similar for both ensembles. I was believed that the reason behind this agreement is the microscopic control of the temperature of each particle done by the langevin thermostat.
Indeed, they have. The problem in my opinion is the fluctuations. I am worried that this brakes the ergodicity of the system. The ultimate goal is to simulate the migration/recombination of this and larer defects so if by removing some long range interactions I truncate the space of possible configurations, the approach is not ok.
Yes. What you are seeing in my graph is obtained by a moving average with a window size of 0.3 ps (My dt is 3e-3 ps, the output frequency is every 10 steps and the smoothing window size is 10 thermo output steps).
Thank you for this suggestion. I think it is the idea that I was following until now, only I didn’t know the name of the model. So far I did the following:
I relaxed the entire system at temperature Ttarget.
I minimized the system keeping the box size fixed
I replaced the minimized mobile atoms by the unminimized ones.
I excluded the inmobile-inmobile interactions and added the tethering force.
I generated the velocities using bolztmann distribution to Ttarget
fix langevin all + fix nve
run
I understand that, in this model, the time-averaged displacement of the background would simply be the amplitude of the oscillations of the tethered atoms.
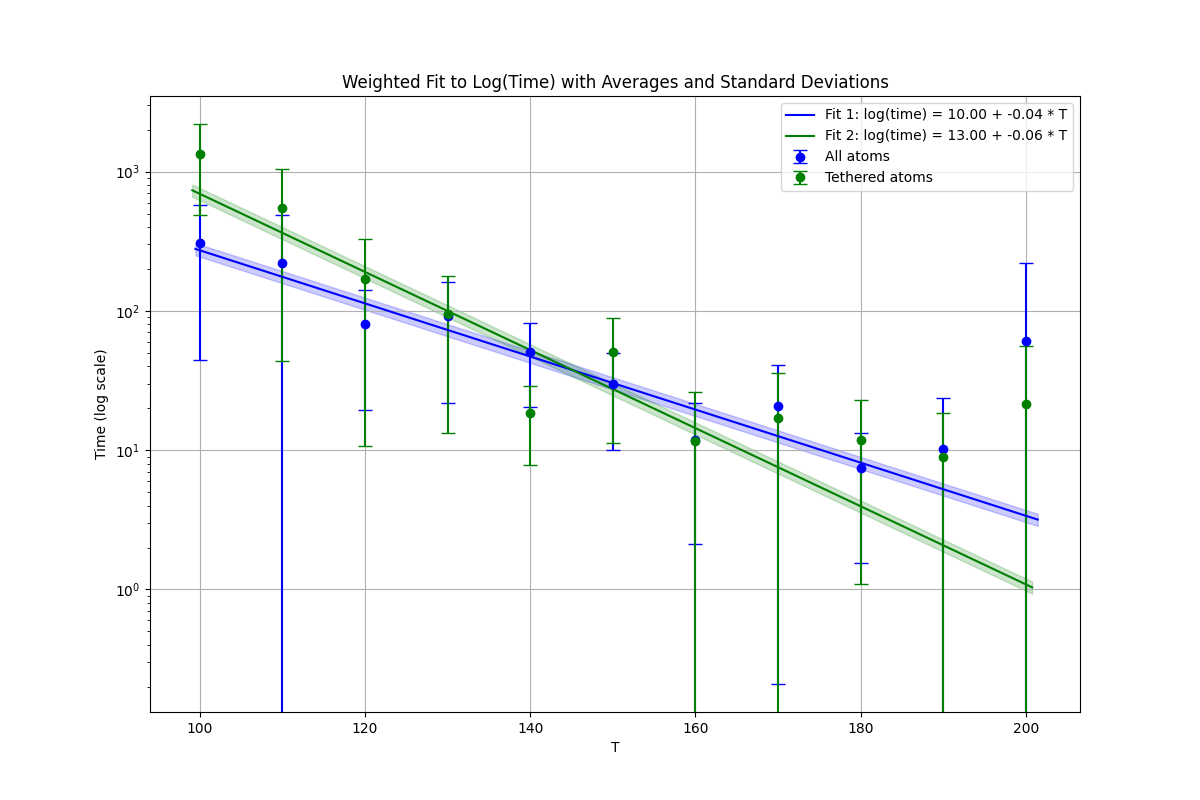
Yes, this is what I am attempting now. In addition to this pressure bias, I attempted to compute the arhenius for the migration of the defect I showed.
The slope is a bit smaller but the uncertainty of the data is large. I am trying to further improve the agreement by examining the pressures, hence my original question.
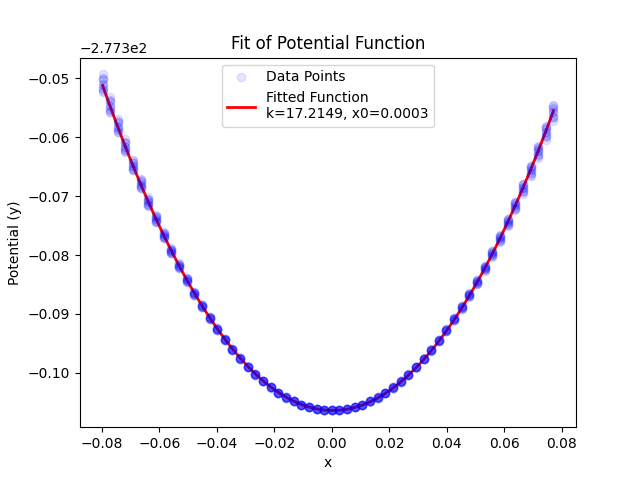
Yes. I tuned the spring constant by performing a potential bassin exploration at different slightly rattled configurations. The amplitude spawns approximately the average KE at room temperature. To this data I fit an harmonic function from which I get the spring constant.
This conversation is becoming really tricky as I can’t tell from the discussion what you have actually done, in turn because there is some imprecision in the terms you’re using, for example:
The imprecision arises since all moving particles must be time integrated (otherwise they wouldn’t move), so “tethered” is independent of “time integrated”. In turn, on your original graph, you had the blue curve as “excluding atoms from neighbour list” which is also independent of either time integration or tethering.
These are three separate interventions after a thermal equilibration and an energy minimization (which seems to me to cancel out the purpose of thermal equilibration) and then swapping out some positions with others and then excluding neighbouring interactions for … some, but not all, particles? And then regenerating random velocities?
Those are many steps, and the more steps you have, the more chance you have that:
the commands for any one step might have some error or typo, making it do something different from what you intended
the effects of any two or more steps contradict each other
even if all steps are correctly specified and coherent, communicating those steps to another reader will run into issues.
From my current understanding of your procedure, you’ve replaced a background of interacting atoms with non-interacting atoms; a lattice of non-interacting atoms is trivially much “softer” and thus, as a pressure reservoir, permits much higher pressure variations in your subsystem of interest. But I am not sure I quite understand what you are doing.
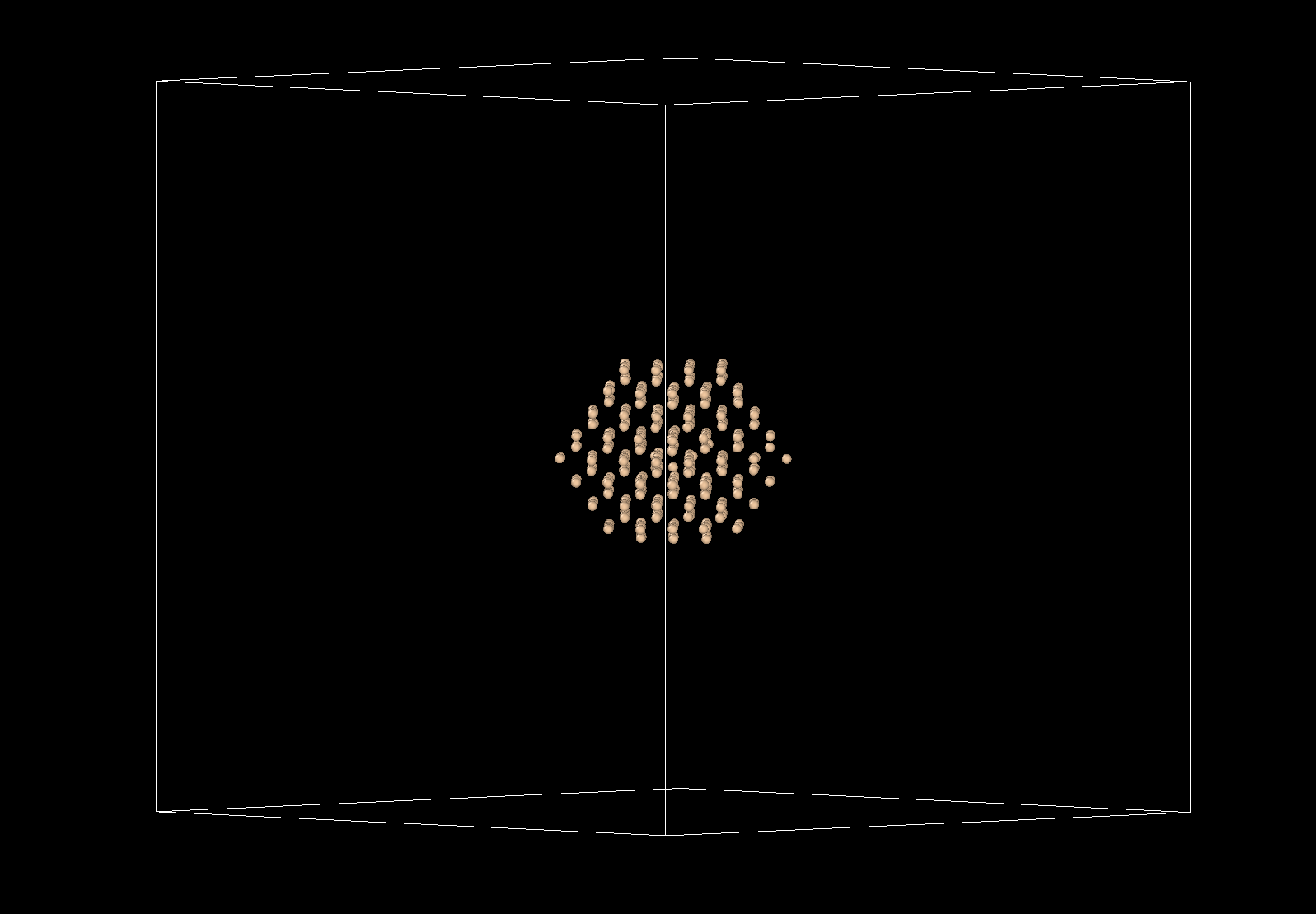
Sorry for that. Maybe I can explain a bit better. I am trying to simulate the long term dynamics of defects in solids. An example for diamond is shown below:
In red you have the only defect: a self interstitial for which I am interested to compute e.g the migration time. I am also planning to use the same idea for extended defects or even amorphous regions.
My assumption is that, to simulate the dynamics of the particle of interest (in red) I could prescind from the atoms that are far away enough from it. I thought that this assumption is the same than when we use finite size boxes in our simulations. Following this assumption I only included pair interactions for the particles within say 2 nm of the defect (those are the particles in blue). For the yellow particles the only force that acts on each is a spring that bounds them to their original position. The constant of this spring simulates the potential energy well in the vicinity of the local minimum (perfect diamond lattice).
I understand that the idea is complicated but I think my steps make sense, don’t they? The all atom relaxation equilibrates all particles, red+blue+yellow so that red and blue are properly relaxed. The embedding (yellow) particles are minimized so that x0 in the tethering force is the original position and not a random non-zero temperature position. This is the step that gives me the right average pressure.
Indeed, but in my case there is an interaction. The particles are tethered to their original position by a spring-force.
I still can’t really follow. I think the most productive thing to do is to post your LAMMPS script here.
Please note that if we can determine that LAMMPS is in fact doing what your script does, then that’s the main purpose of this forum served. Trying to determine if the results are scientifically correct is ultimately the responsibility of users and co-authors.