Hi Lammps users and developers
I want to equilibrate a crystal structure and studied the most descriptions in
mailing list about equilibrium. special thanks from Axel and Steve to good descriptions about the equilibrium state in mailing list.
I created the unit cell, minimized it, replicated (3840 atoms create) and then run equilibrium phase of my simulation for 50 picoseconds by time step=0.001 (metal units) with these commands:
velocity all create 300 234 dist gaussian
fix 1 all nve
fix 2 all langevin 300 300 1 48279
after that I unfix the langevin and just run for the other 50 picoseconds to test my equilibration state.
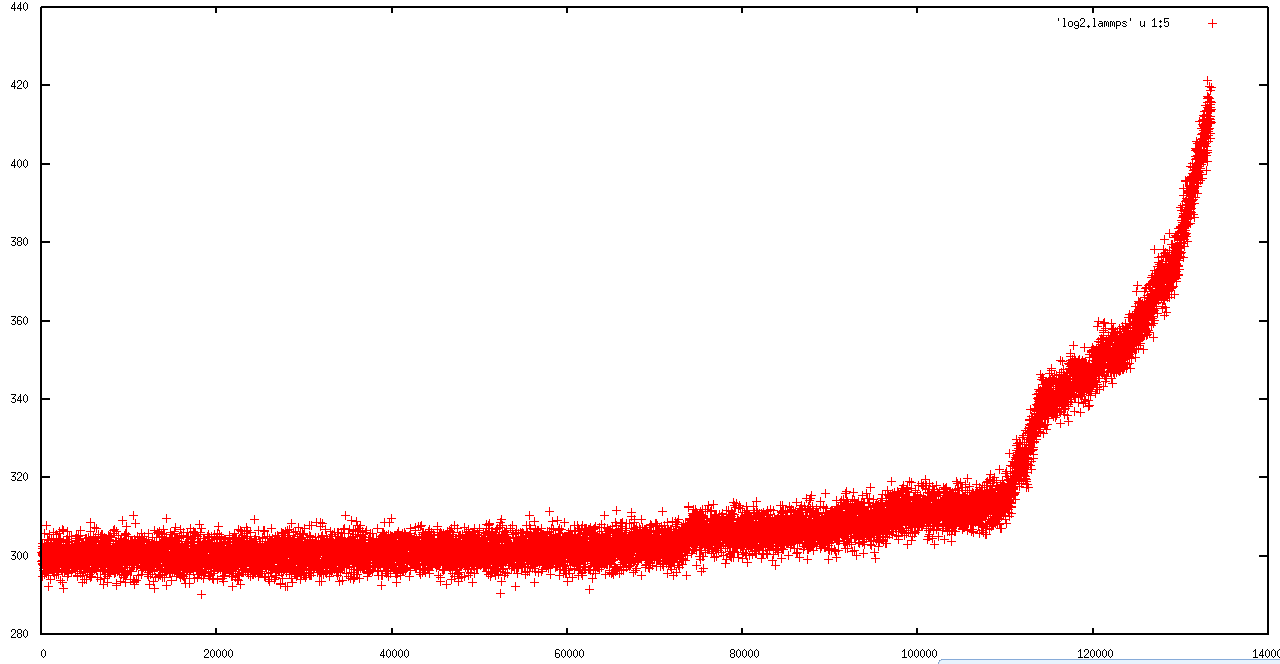
in first 50 ps, everything was ok ( I am not sure), but in second run the simulation ran just for 130000 steps (130 ps) and then the temperature rise and the "error: lost atoms " occur.
please advise me where I am wrong. I am worried because in next steps of my simulation I should arise the number of atoms to about 100000 atoms and use P P S boundary condition and interact whit other material.
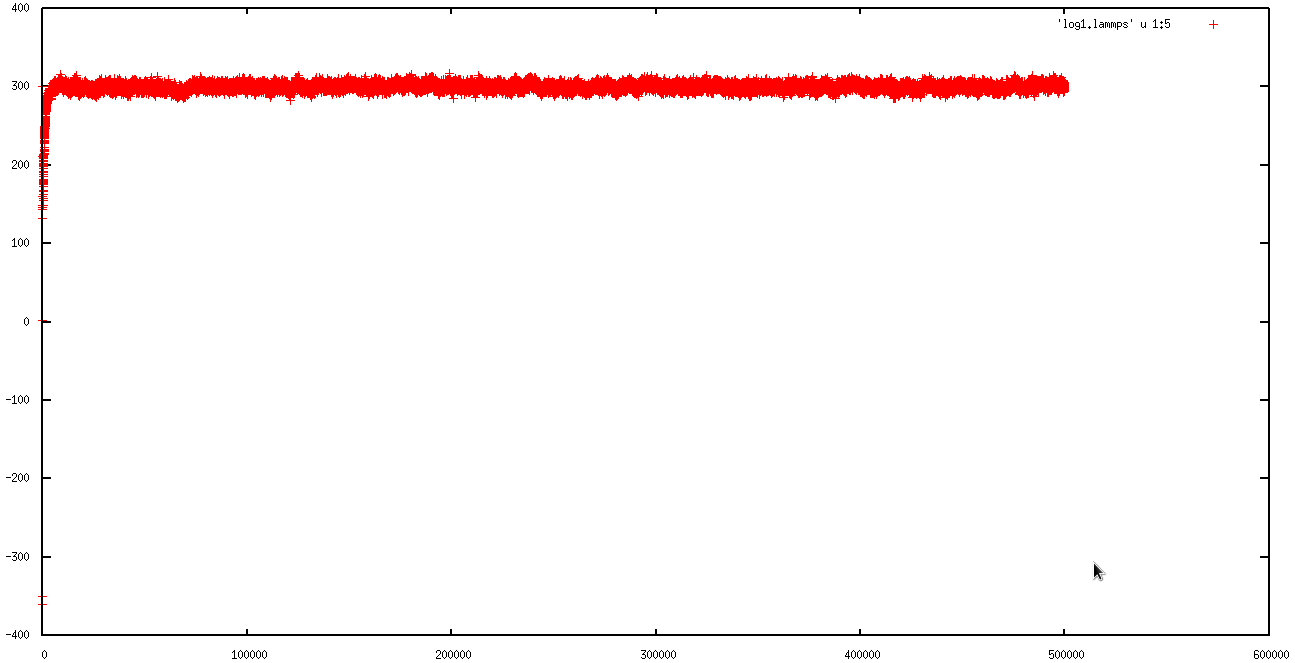
I attached the behavior of MSD, Temperature and total energy in first and second runs to explain the problem better.
Thanks in advance
Mohammad Ashajer
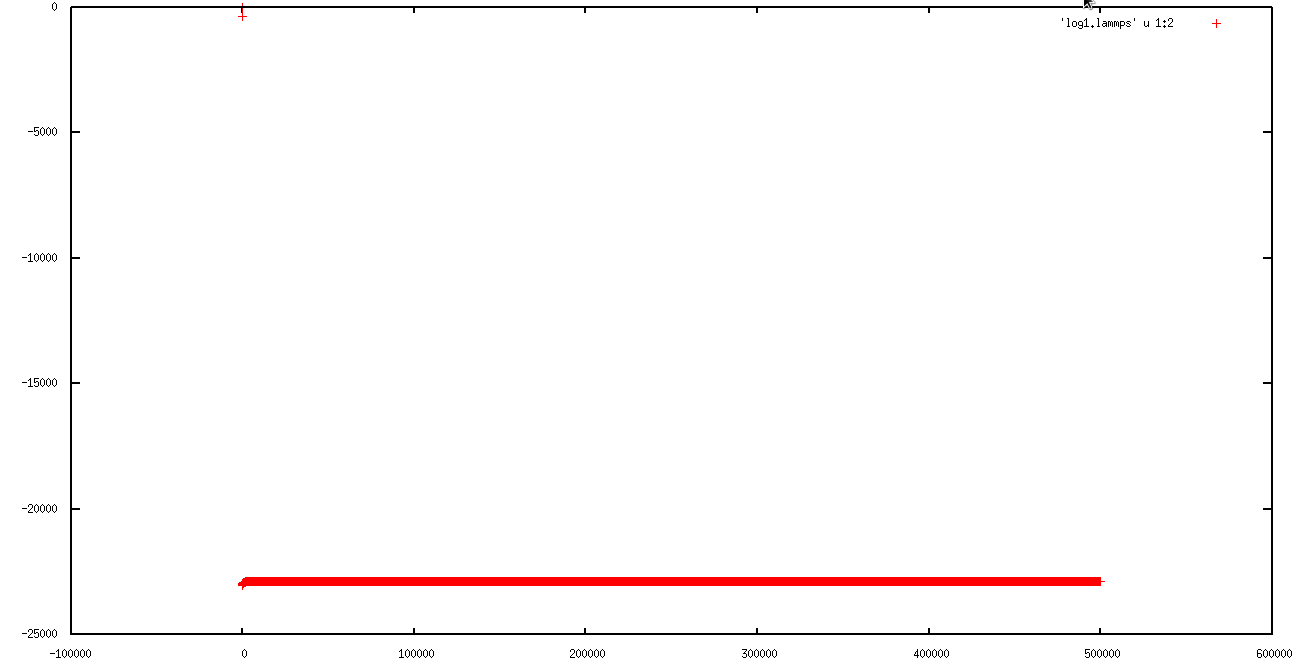
![first_run_msd[4].png](/uploads/default/original/2X/3/3b2115cad5fceedff3d189ba2d737cb86c75142b.png)
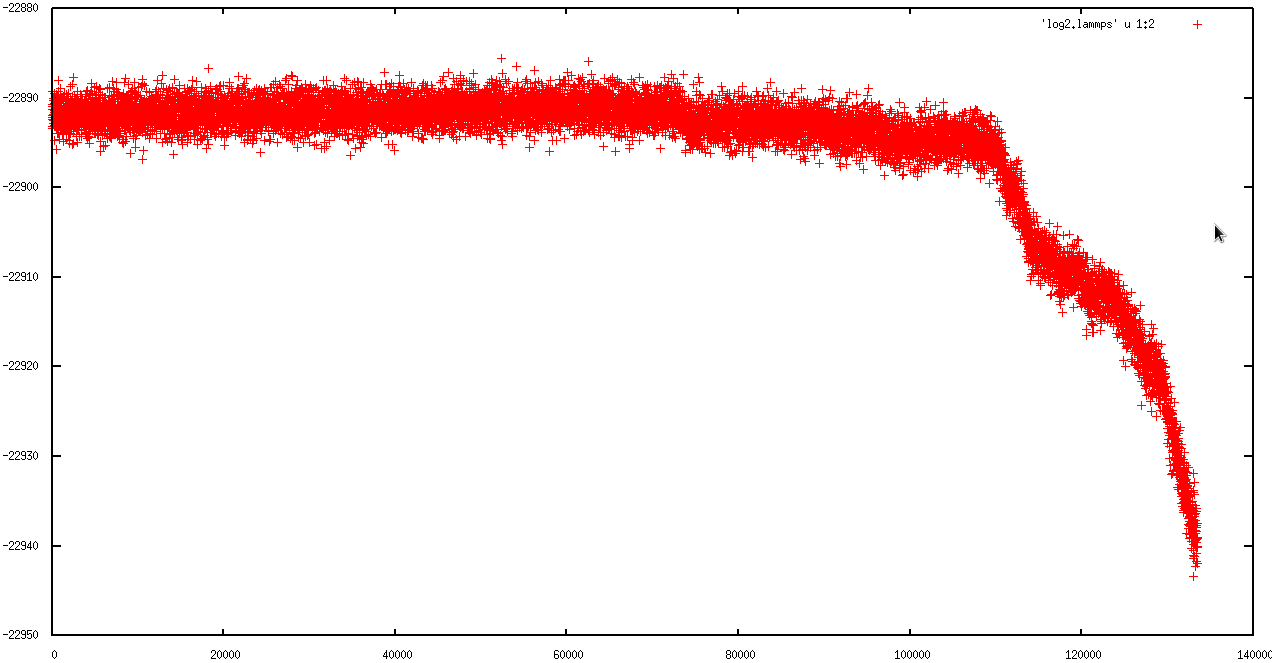
![second_run_MSD[4].png](/uploads/default/original/2X/7/7f63e408c39a76759cea35722656d16bb6bc4444.png)
Send your full input file if you want people to be able to help you. Otherwise send your message to a clairvoyant list where mind reading is performed.
Carlos
Dear Carlos
thanks for your attention and reply.
This is the input script.
 SIm-Input.tar.gz
SIm-Input.tar.gz
the files are not made public
the msd show that the langvin thermostat (in my simulation) make the atoms move very much.
then I use nvt insted of nve&langvin. it make the msd goes better but finaly the system make error and diverge in second run.
Thanks in advance
Mohammad ashajer
Mohammad,
You should add box relaxation to the minimization strategy as well to make sure your cell ends up having the right shape the potential requires and no initial stresses of any kind exist. If the problem that you observe (nve+langevin vs nvt) remains, then chances are you might be working with a defective potential and I would suggest to try reproducing the original results the authors of the potential published when they came up with this paramterization. You could have set the wrong parameters in the meam file.
Carlos
Dear Carlos
I added box relaxation and simulate with nve+langevin, but the simulation stop after 200000
step in first step. I do it for nvt and then reached the same result as nve+langevin. therefore, result show added the “box relaxation” was not good for this situation. (I don’t know why)
I do the simulation with initial temperature ( velocity all create 300 234 dist gaussian) without any thermostat (only “fix nve”). after a few steps the temperature come down to 150k and this continued for 1000000 steps. I did the simulation again with “box relaxation” and the temperature come down to 150 k and continued. I expect the simulation should be diverge without thermostat, but the results show it diverge with thermostat.
1- is there any advise why this happened?
2- I plan to do the simulation with initial temperature about 600 k and it come down about 300 k
and continue to 1000000 steps. please let me know your suggestion about this method to
equilibrate the system?
Thanks in advance
Mohammad
Mohammad,
You need to try to understand why that is happening. I don’t have the time to dig into the details of your project. Your input script has no obvious mistakes related to Lammps but I have no idea how well is your aluminum oxide implementation of the potential.
My advice as already stated is to try reproducing the results of the original authors in order to properly test the potential. A combination of fix nve+langevin should not destroy your oxide structure. Your *.meam file cites as reference the rocksalt b1 structure and includes a cohesive energy and first neighbor equilibrium distance for it. Have you tested that you can indeed reproduce those numbers if you built such a lattice? Use common sense and look for precise benchmark examples for this potential to compare your numbers to.
Good luck,
Carlos