I’ve been using LAMMPS for about three years now and have generally been able to resolve issues by consulting the documentation or previous forum discussions. However, I’m currently facing a problem that I can’t seem to solve despite my best efforts.
Here are the details of my issue:
LAMMPS Version : LAMMPS (23 Jun 2022 - Update 4)
Context : I used a Python script to generate an initial polymer data file (attached as “wall.txt”) using the LAMMPS format (with atom_style full, including the Atoms and Bonds sections). This polymer is intended to interact with other particles that I later introduce using the create_atoms command. My simulations are coarse-grained, and the scripts provided are simplified examples, not representing realistic systems, but they effectively illustrate the issue I am encountering.
Problem Observed : The atoms read from the wall.txt data file do not respect the pairwise interactions defined in my LAMMPS script (lj/cut to simulate WCA potential). Specifically:
Interactions between “atoms generated with create_atoms” and “atoms from the data file” behave as expected.
Interactions between atoms generated by create_atoms also behave correctly, repelling each other as intended.
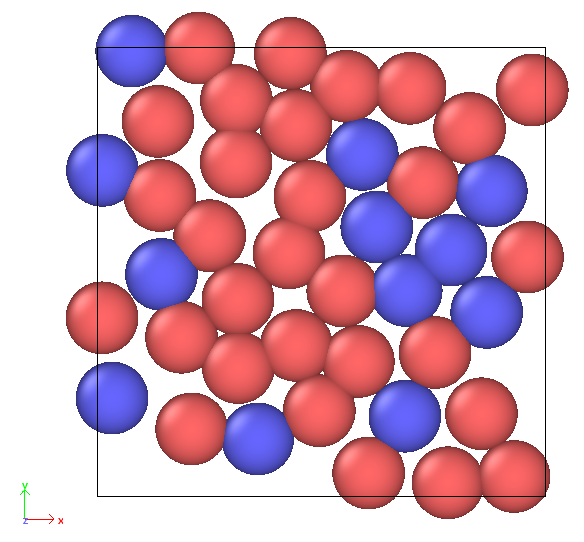
However, interactions between atoms within the wall.txt data file are ignored, leading to unexpected overlap. (cf. the Ovito snapshot: atom type 4 is represented in pink. We can observe the upper pink atoms repelling each other, whereas the lower pink atoms are overlapping.)
What I Have Tried :
Since the wall mainly consists of atoms of type 4, I attempted to create additional atoms of type 4 using create_atoms. Interestingly, these newly created atoms behaved normally (i.e., they repelled each other as expected), while the original type 4 atoms from the polymer data still overlapped.
I then attempted to “reset” the system by writing a new data file using write_data in an initial script (see “test1.lmp”) and reading it again in a subsequent script (see “test2.lmp”). My hope was that this would erase any information from the problematic read_data stage. Despite this, even with a fresh data file created through write_data—where no distinction should exist between atoms of type 4 in the Atoms section—the polymer atoms continued to overlap, while the newly created type 4 atoms still repelled one another.
This doesn’t make sense.
Once atoms are created, LAMMPS doesn’t know how they were created.
I do notice that the data file has some atoms of type 3 and some of type 4, while your description seems to imply that all atoms in the data file are of type 4. Since the epsilon for interactions with atoms of type 3 with each other is set to 0, it seems that the problem is not a LAMMPS problem, but is located between the seat of your chair and your keyboard.
The data file does indeed contain atom types 3, 4, and 5. I mentioned only atom type 4 to focus on the core of the issue. The atoms of type 3 are meant to be frozen, which is why the epsilon is set to 0 (they represent the yellow substrate in the Ovito snapshot). However, I don’t believe this is related to the overlap problem I observe with atom types 4 and 5 from wall.txt.
Please understand, that I am not going to dig through your input (considering that I already have a hard time following your logic describing it) to do your debugging for you.
You can do the debugging on your own, by starting with an empty input and then just adding bits and pieces in stages and observing carefully what they do and if this is in accord with the documentation.
Or you can produce a minimal input deck (with just a few atoms) that showcases your problem and thus can be far more easily debugged and understood by people not familiar with the intricacies of your project.
You have bonded atoms with a harmonic bonds model and a force constant of 80 and an equilibrium distance of 1, particles that also repel one another with a Lennard-jones potential that has parameters \varepsilon=10000 (!!!) and \sigma=1. They also start very close to one another with a distance of 0.25
The model does not seem very reasonable to me, and I think a problem in the code is not the main source of concern here (you should still consider updating.).
Also note that this does not simulate a WCA potential since you end up with a positive force at the cutoff. The WCA potential cuts out at 2^{1/6}\sigma, not at \sigma.
Dr. Clavier, as mentioned in my initial message, “the scripts provided are simplified examples and do not represent realistic systems.” I intentionally used exaggerated potentials to highlight the issue, but under normal circumstances, I typically use FENE potentials with more reasonable coefficients. Nonetheless, I appreciate your feedback on this.
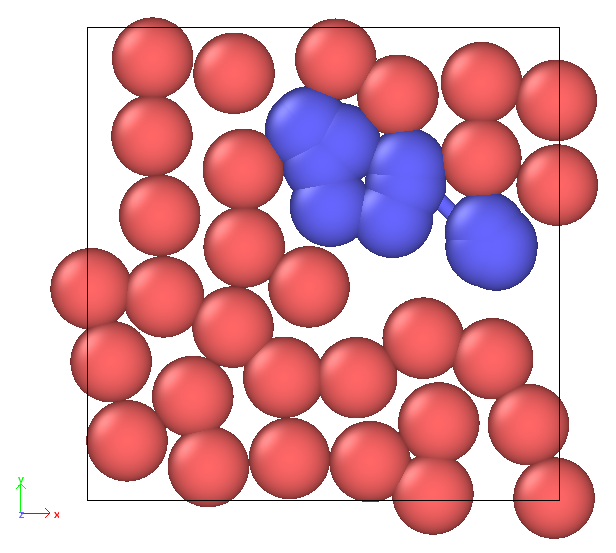
Prof. Kohlmeyer, you are absolutely right—I should have started with a simpler minimal example. Doing so has indeed helped me better identify the source of the issue. The problem seems to arise specifically when I define the bonds in the data file.
When bonds are included in the data file that LAMMPS reads via read_data, the bonded atoms appear to ignore pairwise interactions and overlap, even though the bond interactions themselves seem to function as expected. However, when I remove these bonds from the data file, the particles behave as anticipated, repelling each other in accordance with the pairwise lj/cut settings. I have attached two minimal inputs that illustrate these different behaviors. The problem is likely due to the way I write the data file, but I am still unsure what I am doing wrong. I also tried using different atom styles (such as angle) in the data file, but encountered the same issue.
This is the expected behavior for molecular force fields. It would otherwise make the parameterization of bonded interactions extremely difficult, since they need to be linked to the atom types of the participating atoms. If you read up on these so-called exclusions, you will learn those are usually applied to direct neighbors (1-2 neighbors), neighbors of neighbors (1-3 neighbors) and sometimes even one level more (1-4 neighbors). How those exclusions are applied is controlled in LAMMPS by the “special_bonds” command.
For bond style fene, you are usually required to use the corresponding special_bonds setting. Even for special_bonds fene, the 1-2 non-bonded interactions are excluded.
No worries. Still, theses files were actually different compared the problem you described. As Axel mentioned, part of the issue apparently stemmed from the use of bonds and the special_bonds default settings. Then noting there inclusion/exclusion in the actual file was an important difference.
Thank you both for your guidance throughout this thread. The use of special_bonds lj 1 1 1 has indeed resolved my issue. Thank you for the time you spent helping me understand this.