Dear All,
I used a 25 Å water droplet on a graphene surface locating at z=0. My problem is the strange equilibration procedure which the droplet moves upward (from its initial location) and not relaxed on the surface.
My procedure:
First of all. I double-checked and there were no force acting on the droplet. I used below commands for my case;
pair_style lj/cut/coul/long 12.0
pair_coeff 1 1 0.0700 3.9848 #C-C
pair_coeff 2 2 0.0 0.0 #H-H
pair_coeff 3 3 0.1553 3.166 #O-O
pair_coeff 1 2 0.0 1.9924 #H-C
pair_coeff 1 3 0.1043 3.5754 #O-C
pair_coeff 2 3 0.0 0.0 #H-O
kspace_style pppm 1.0e-5
bond_style harmonic
velocity carbon set 0 0 0 units box
fix NOFORCE1 carbon setforce 0 0 0
velocity water set 0 0 0 units box
fix NOFORCE2 water setforce 0 0 0
So, I used the VMD program to visualize dump.xyz output file. But I do not understand why the surface is located at zmax instead z=0 (droplet also locates below surface correspondingly). by this way, every things looks freeze without movement…
In the second place, I added NVT ensemble for the water molecule and it got worse. Water molecules starts to move from their initial place on the surface to the z=0 of the simulation box.
fix NVT water nvt temp 298.0 298.0 100
The LJ parameters are also double-checked for any mistakes.
Any comments are most welcome,
There have been discussions on simulating water droplets before. I suggest you look them up through the forum search feature or a google search.
The information you provide is insufficient to see and reproduce what you claim you see. Without that, it is impossible to provide meaningful comments. What you see is likely caused by what you don’t show and tell us.
Thanks for your comments @akohlmey
I reviewed almost all topics regarding droplet modeling on the solid surface and hereby found my above problem is resolved with fix wall/reflect command. Now, I cannot imagine why I get lost atom error.
To solve this problem:
- I decreased time steps into 0.5 fs. But, it is not working.
- I tried with
fix momentum. But, again I get the similar error exactly at the same running time.
- If I undo the fix shake command, I get no error strangely!
I share the input file for any comments.
Any comments are most welcome,
wetting.in (1.4 KB)
wetting.data (958.6 KB)
There is a major blunder in your input. SPC/E is a rigid water potential, but the H-O-H angle is not defined in your input and not held rigid with fix shake. That can lead to all kinds of unphysical behavior. It should be clearly visible when visualizing your system.
There are many other things that I would do differently:
- there is no need to compute the carbon-carbon forces
- I would use the processors and the balance command to improve performance
- I would use a more relaxed kspace convergence (you have a fixed box so pressure contributions do not need to be fully converged).
- I would only time integrate the water atoms and monitor the temperature of those atoms only from the temperature compute created and used by fix nvt
- I would split the equilibration into two parts, one with a ramp, the other with a constant temperature.
- I would use the velocity command to zero the translational momentum after the two equilibration runs each.
- I would use compute momentum to monitor the momentum of the water atoms
- I would use fix shake already during minimization and use bond and angle style zero for simplicty and efficiency
- I would remove all the carbon-carbon bonds from the data file (those atoms won’t be allowed to move anyway).
- I would set the charges directly in the data file
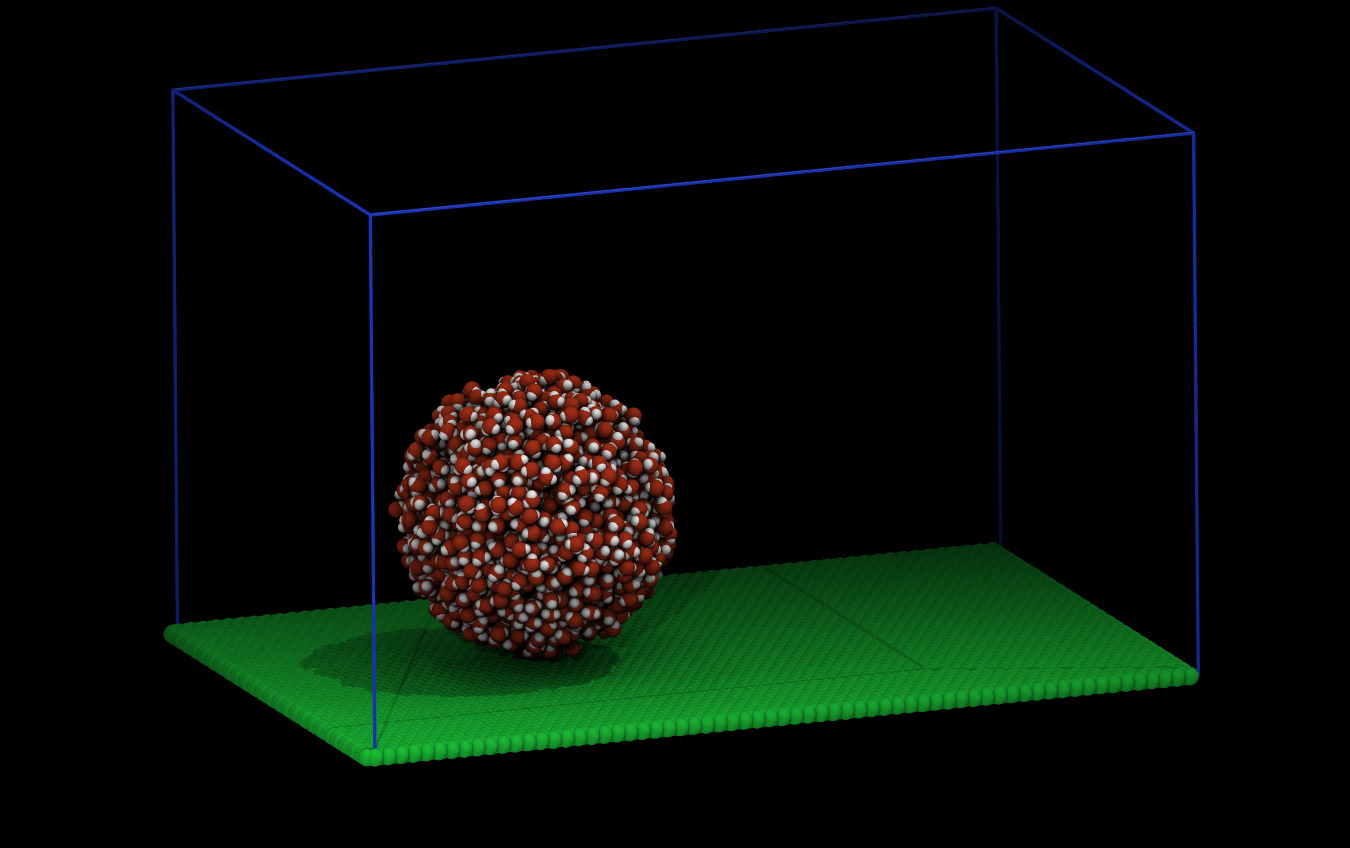
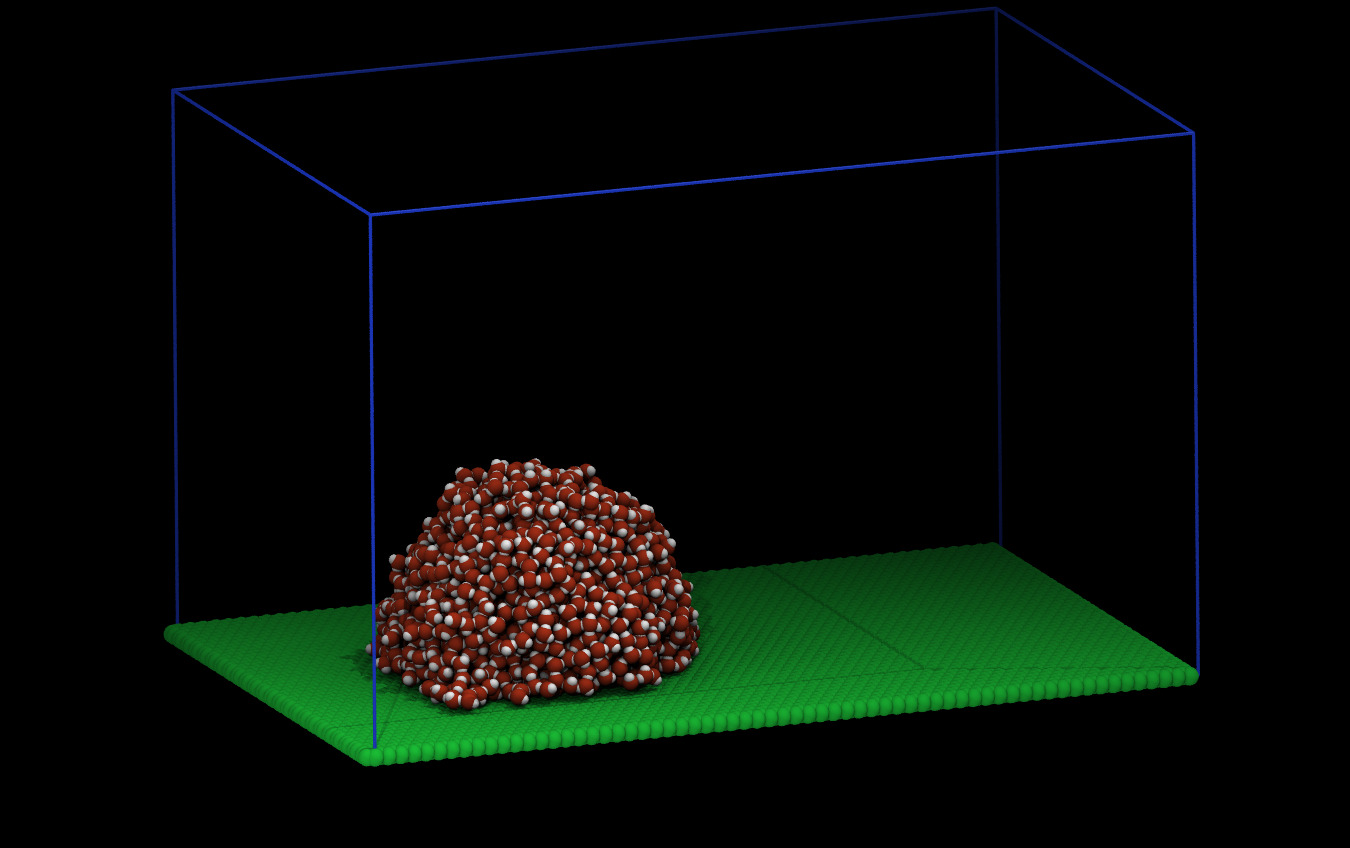
Here is what the system looks like for me at the beginning and after (some) equilibration:
Here is the corresponding log file.
log.droplet.gz (30.7 KB)
1 Like
Now it works. Thank you very much @akohlmey for the time and energy you put in yours to promote our understanding.
And it’s unclear for me about using fix momentum in order to restrict droplet movements on the surface during these two equilibrium.
So, should fix momentum be applied into just x, y directions or into all three directions?
For me, I applied it into just x and y direction. Then for after equilibrium process, just unfix it and use velocity set 0.0 0.0 NULL But I do not know is it correct or not
All of these are comments and questions about the science of what you are doing and not LAMMPS questions. You need to discuss this with the people that advise you on the science of your work.
Besides, you cannot write in your publication “I chose this option because some dude in the forum told me so”. You need to justify your choices with proper scientific arguments. LAMMPS just provides the tools and will work as documented regardless of the scientific meaning of the choices you make.
In other words, the forum cannot fill in for your adviser.