Dear LAMMPS users,
suppose that the lattice structure of your material is monoclinic such as SiC, SiN and etc. However, you have to do your simulation on a cubic simulation box. For the sake of thism I used the following code, define a cubic region and fill it with atoms as follow:
units metal
atom_style atomic
dimension 3
boundary p p p
#------------Define variable------------------------
variable n equal 10
variable a equal 7.5572
variable b equal 7.5572
variable c equal 2.8852
variable gamma equal “120.0030/180PI"
variable singamma equal “sin(v_gamma)”
variable cosgamma equal “cos(v_gamma)”
variable bx equal "v_bv_cosgamma”
variable by equal “v_bv_singamma"
variable prismx equal "v_bv_cosgamma/v_n”
#------------Define Si3N4 lattice------------------------
lattice custom 1 &
a1 a 0.0 0.0 &
a2 {bx} ${by} 0.0 &
a3 0 0.0 $c &
basis 0.1742 0.7678 0.2500 &
basis 0.2322 0.4064 0.2500 &
basis 0.5936 0.8258 0.2500 &
basis 0.8258 0.2322 0.7500 &
basis 0.7678 0.5936 0.7500 &
basis 0.4064 0.1742 0.7500 &
basis 0.3333 0.6667 0.2500 &
basis 0.6667 0.3333 0.7500 &
basis 0.3302 0.0299 0.2500 &
basis 0.9701 0.3003 0.2500 &
basis 0.6997 0.6698 0.2500 &
basis 0.6698 0.9701 0.7500 &
basis 0.0299 0.6997 0.7500 &
basis 0.3003 0.3302 0.7500 &
orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
#------------------Create SiC nanowire--------------
region zona1 block 0 75 0 75 0 15 units box
create_box 2 zona1
create_atoms 1 box &
basis 1 2 &
basis 2 2 &
basis 3 2 &
basis 4 2 &
basis 5 2 &
basis 6 2 &
basis 7 1 &
basis 8 1 &
basis 9 1 &
basis 10 1 &
basis 11 1 &
basis 12 1 &
basis 13 1 &
basis 14 1 &
write_data SiC.in
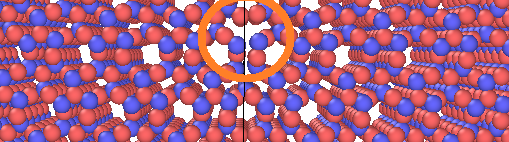
So, my problem is that the structure doesn’t look correct for the sake of periodicity. I attached an image of it and circle around the section of the boundary that as I switch on show periodic image in Ovito, it doesn’t look correct. I mean the structure doesn’t look uniform as we go from left of the boundary to the right of it.