Hello Lammps users and developers,
I am trying to simulate the evaporation of liquid Ar on a flat copper
surface at T=140K. The solid surface contains 9 layers of copper
atoms. The first layer of Copper atoms stayed as a boundary wall to
keep the volume of the system constant (Group S1); the following
inside two layers were set as heat source from which heat flux was
generated (Group S2); the last six layers were set as solid walls
through which heat conducted to argon fluids (Group S3). The group
"SOLID" and "liquid", "Vapor" and "LV" contain all solid atoms,liquid
atoms, vapor atoms and liquid+vapor atoms, respectively,
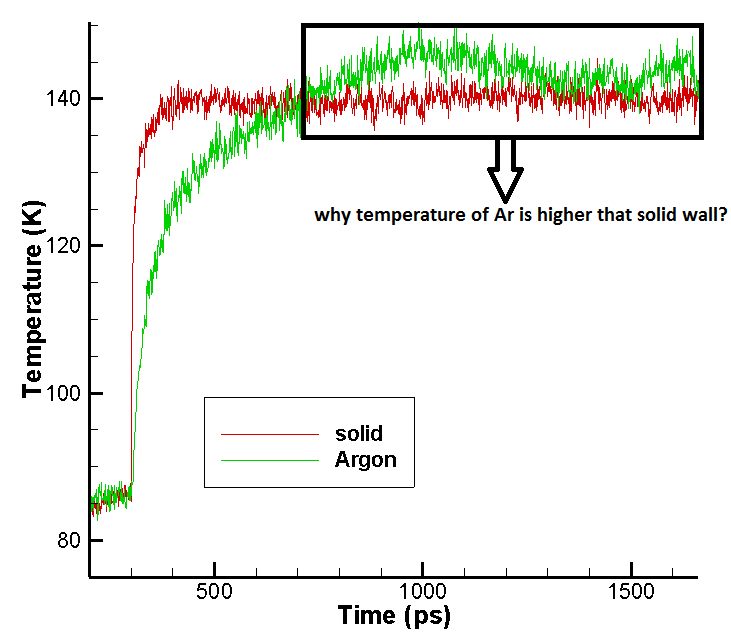
The temperature history during equlibration period is O.K and all the
system is in T=90K but in Evaporation period the temperature of Ar
liquid (T_LV) increases (more than 150 K) and get higher than the
temperature of solid surface (temperature of group 3) which is not
realistic. Would you please help me what is wrong in my input script
and simulation set up?
units metal
dimension 3
boundary p sf p
atom_style atomic
neighbor 2 bin
neigh_modify delay 5
region box block 0 45 0 600 0 45 units box ### simulation box
create_box 2 box
lattice fcc 3.61 ### lattice for solid region
region 1 block 0 45 0 1 0 45 units box ### fix solid atoms region
region 2 block 0 45 1 4.9 0 45 units box ### The region above
the first layer (second region)
region 3 block 0 45 4.9 15 0 45 units box ### last layers of solid wall
create_atoms 1 region 1
create_atoms 1 region 2
create_atoms 1 region 3
lattice none
lattice fcc 5.76 ### lattice for liquid region
region 4 block 0 45 15 30 0 45 units box ### liquid atoms region
create_atoms 2 region 4
lattice none
lattice fcc 32.86 ### lattice for vapor region
region 5 block 0 45 30 600 0 45 units box ### vapor atoms region
create_atoms 2 region 5
lattice none
group S1 region 1
group S2 region 2
group S3 region 3
group liquid region 4
group vapor region 5
group LV union vapor liquid ### liquid+vapor atoms
group solid union S1 S2 S3
group whole1 subtract all S1 ### all atoms-wall_fix
group whole2 subtract all S2 ### all atoms - S2
mass 1 63.5463 ### mass of solid atoms
mass 2 39.948 ### mass of liquid and vapor atoms
set group solid type 1
set group LV type 2
pair_style lj/cut 12
pair_coeff 1 1 0.409598855 2.34 12 ### S-S (free layer with itself)
pair_coeff 2 2 0.0104233 3.40 12 ### L-L, L-V and V-V
pair_coeff 1 2 0.065007161 2.8689 12 ### L-S and V-S(free layers)
compute T3 S3 temp
compute_modify T3 dynamic yes
compute T_LV LV temp
compute_modify T_LV dynamic yes
min_style cg
minimize 1.0e-6 1.0e-8 10000 10000
reset_timestep 0
velocity S1 set 0.0 0.0 0.0 units box
velocity whole1 create 90 482748 units box
fix 1 S1 setforce 0.0 0.0 0.0
fix 2 all nve
fix 3 solid langevin 90 90 100.0 699483
fix 4 LV langevin 90 90 100.0 699483
fix 6 all wall/reflect yhi EDGE units box
thermo 1000
thermo_style custom step c_T3 c_T_LV
thermo_modify line one lost error norm no
dump 1 all atom 500 Cu-flat.vmd
timestep 0.001
run 100000
############# Equlibration Period#################
unfix 3
unfix 4
fix 5 S2 langevin 90 90 100.0 699483
run 200000
############# Evaporation Period#################
unfix 2
unfix 5
fix 8 S2 nvt temp 140 140 0.1
fix 9 whole2 nve
run 2000000
{kind=link}
{kind=link}
{kind=link}