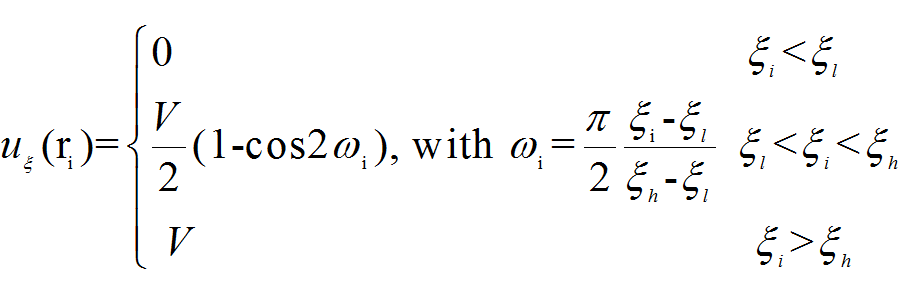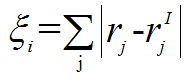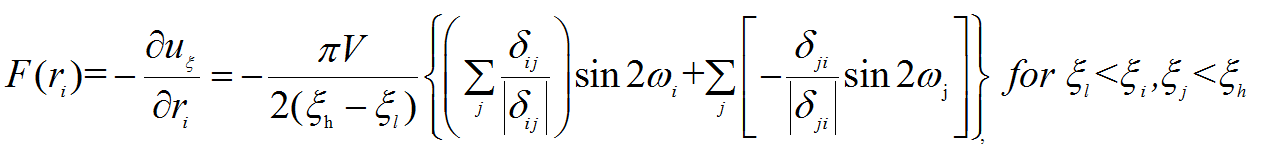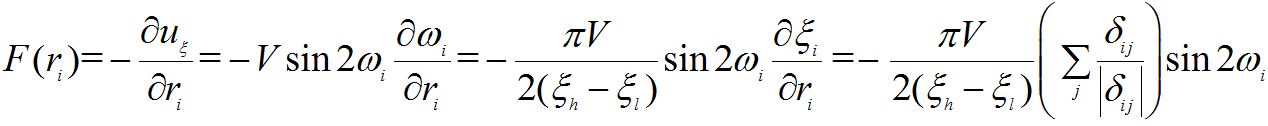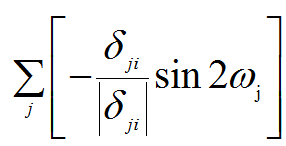I am using the ‘fix orient/fcc’ to calculate GB mobility of fcc material.
The fix adds an orientation-dependent potential energy to atoms (pe.png),
where (order.png)is the order parameter of each atom i. (For more details,
please read the manual and the publicattion(Nature Mater 2006,5:124-7)).
Then by evaluating the derivative of the artificially added potential energy, we can
get the additional force on each atom (deriva.png),
While I have some doubts about the above derivative, in my opinion, the derivative should be (deriva_me.png),
i.e. without (deriva_part.png). Is it right?
I think it should be my wrong understanding of the derivative that leds to my doubts.
However, I still want to know how to specifically get the derivative as given in the publication.
PS:If figures of function inserted in this email cann’t be displayed, please enter this email.
user:yangliang_lammps@…543…
password:yangliang123
Any comments from you will be sincerely appreciated.
PS:If figures of function inserted in this email cann't be displayed,
please enter this email.
user:[email protected]...
password:yangliang123
congratulations. you just announced to the entire online world how your
e-mail account can be hacked.
as if there aren't enough compromised e-mail accounts spewing out spam
already.
I have sent emails to more than one lead authors about
a month ago, but I have not received any reply. So I have
to turn to lammps-users for help.
I know my problem is silly for knowledgeable people. I
have read some data about derivative of discrete function,
but I still can not figure out how to get the derivative
given in that publication.
It’s fine if you want to send to the mail list.
I just think it unlikely that anyone here will dig into
the techincal issues you ask about, deep enough
to give a good answer.
 (pe.png),
(pe.png), (order.png)is the order parameter of each atom i. (For more details,
(order.png)is the order parameter of each atom i. (For more details, (deriva.png),
(deriva.png), (deriva_me.png),
(deriva_me.png), (deriva_part.png). Is it right?
(deriva_part.png). Is it right?