Dear all,
I am trying to make a lammps data file from .car and .mdf file using msi2lmp.exe.
My full journey: I build my monomer using medeA software and saved it as .pdb file. Then, open this .pdb file in Materials Studio and export it as .car file and finally try to convert it to lammps data file along with cvff parameters using msi2lmp.exe. I am using this command “msi2lmp.exe Vitrimer_Monomer -class 1 -frc cvff”.
But getting this error:
Building internal coordinate lists
WARNING inconsistent # of connects on atom 3 type xx
WARNING inconsistent # of connects on atom 4 type xx
WARNING inconsistent # of connects on atom 5 type xx
…
Reading forcefield file
Get force field parameters for this system
Unable to find mass for xx
I am sharing my my .pdb, .car and .mdf files. Please go through the link to see:
https://drive.google.com/drive/folders/1E8QC0gDc0TJP4bqQ8U28UglV-lZ4LjjJ?usp=sharing
Why I am getting this error? Is there anything wrong in the .pdb, .car and .mdf files? If it is then what should be the right way to generate these files or how can I solve the problem?
Thanks.
Because your files assign an atom type “xx” that is not known to the CVFF force field.
Both the .car and the .mdf files are missing information. There is no atom type assignment and the charge group assignment.
A crucial step of force field calculations is to assign force field atom types to individual atoms based on the “rules” for the desired force field. Only then can the msi2lmp tool assign force field parameters for all interactions (assuming that parameters are available for all assigned types and combination of types). This is obviously missing, so you will have to do that and create suitable .car and .mdf files.
In addition your .mdf file contains bonds that are (incorrectly) guessed from a distance search.
They are not present when loading the .pdb file into VMD with “autobonds off”, but appear with “autobonds on”.
Thanks for your reply.
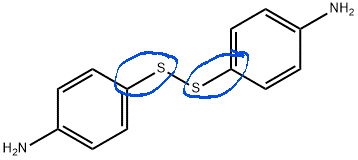
In my system, there is a 4-aminophenyl disulfide (AFD) structure. But after assigning all atom types manually in .car file there is an error “Unable to find bond data for cp s1”. That means there is no bond data for the bond of Sp2 aromatic carbon (partial double bonds) and Sulfur involved in S-S disulfide bond in AFD in the cvff force field.
As there is no bond data for cp and s1, can’t I use cvff force field for my system? Or What can I do to solve this problem?
AFD Structure
Don’t forget that you also have bogus bonds in the .mdf files that need to be removed or avoided.
Not directly unless you can find or derived the additional, missing parameters. There is a little hope as you could manually search through the equivalence tables in the force field file where it says that you may replace missing parameters involving the s1 atom type with those for the s atom type. the msi2lmp tool will not apply those, if I remember correctly.
Please also note that the force field file used by LAMMPS is one containing only public parameters and the latest mentioned publication is from 2002. So there may be additional publications with additional parameters or there could be more parameters in the proprietary versions bundled with materials studio.