Dear all,
I am now simulating the branched polymer, the backbone is ellipsoid, and the sidechain is spherical. And I post my model snapshot below. For each backbone ellipsoid, I added three massless points(the black dots) bonded to the backbone ellipsoid, and then use ‘fix rigid group N’ to control each backbone ellipsoid as an individual rigid body (Treat one ellipsoid and three massless dots as one rigid body). SO that I can capture the orientation behavior of the backbone or pi-pi stacking of the backbone. But there is a problem, LAMMPS only allows < 32 rigid groups and I can not increase my polymer chain repeating unit, let alone the bulk system. Is there any solution to this problem? Thanks in advance. Any response would be appreciated.
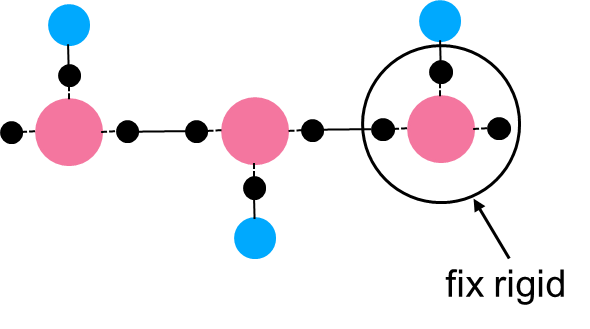
Give each ellipsoid a different molecule ID and then use the “molecule” option of fix rigid to define the rigid bodies. For increased efficiency you probably want to use fix rigid/small where this is mandatory.
Thank you so much, and now it works.
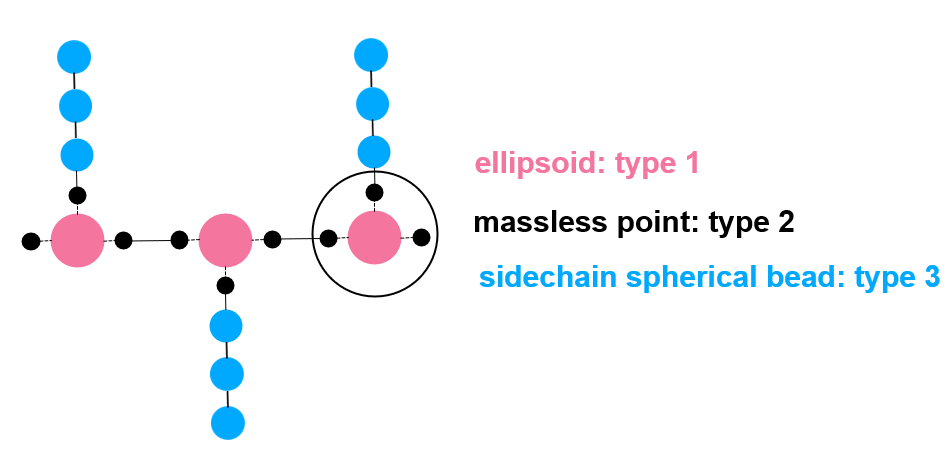
By the way, I have another question about this command. In the picture I shared, one ellipsoid and three massless points are treated as one mol ID, and each side chain (three spherical beads) is treated as another mol ID.
So if I only want the backbone to be rigid, then I should create a group termed backbone that contains all backbone mol IDs, and use “fix 11 backbone rigid/small molecule”. Is that right?
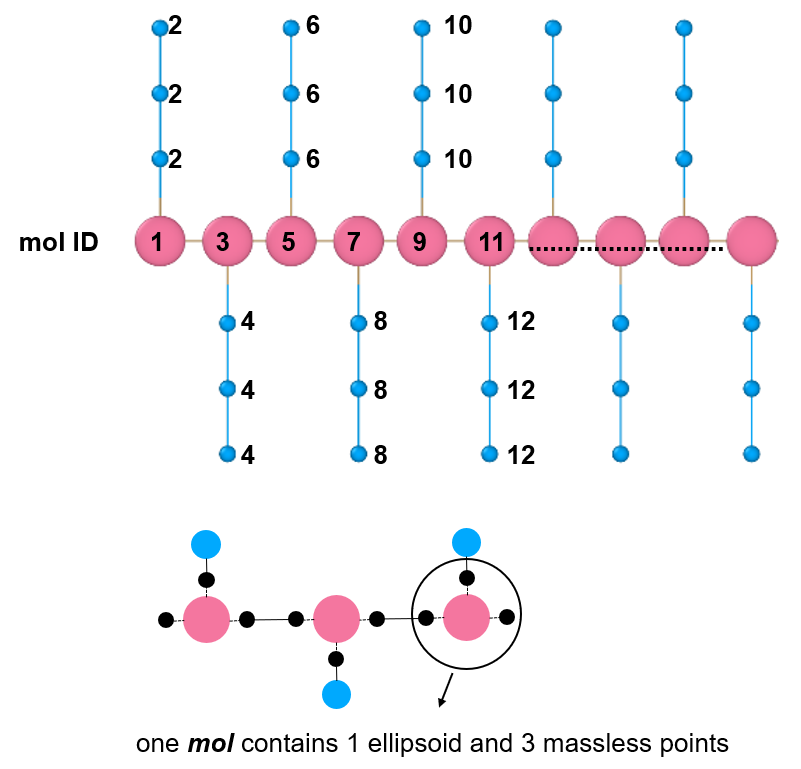
I think so, but that is easy to verify by giving it a try and visualizing the resulting trajectory.
Thank you so much, and I have tried the following two sets of command and the trajectories look reasonable except for the temperature control.
(the backbone group contains all rigid backbone ellipsoids)

First one:
compute myTemp all temp/asphere dof all
compute myPress all pressure myTemp
fix 33 all npt/asphere temp 300 300 400 iso 1 1 4000
fix 11 backbone rigid/small molecule
The temperature is stabilized at around 30 K:
Step TotEng KinEng Temp c_myTemp PotEng
0 59.613798 40.774985 34.283616 34.283616 18.838813
100 59.254057 39.31502 33.056076 33.056076 19.939037
200 57.933463 40.177001 33.78083 33.78083 17.756462
300 56.628364 42.505455 35.738595 35.738595 14.122909
400 55.501463 37.894665 31.861842 31.861842 17.606797
500 55.053838 37.324869 31.382757 31.382757 17.728969
600 54.780034 40.586892 34.125467 34.125467 14.193141
700 55.016761 36.864091 30.995335 30.995335 18.15267
800 54.983528 37.395341 31.44201 31.44201 17.588186
900 55.049312 38.304953 32.206812 32.206812 16.744359
1000 55.441185 39.674831 33.358606 33.358606 15.766353
1100 56.952193 40.540905 34.0868 34.0868 16.411288
1200 58.716594 39.863019 33.516834 33.516834 18.853575
1300 59.850001 40.808491 34.311787 34.311787 19.04151
1400 60.499531 38.11867 32.050185 32.050185 22.380861
Second one:
compute myTemp all temp/asphere dof all
compute myPress all pressure myTemp
fix 33 all nvt/asphere temp 300 300 400
fix 11 backbone rigid/small molecule
This time, the temperature is stabilized at around 2K:
Step TotEng KinEng Temp c_myTemp PotEng
0 -104.09191 2.4312848 2.0442248 2.0442248 -106.52319
100 -104.26142 2.3647407 1.9882744 1.9882744 -106.62616
200 -104.52958 2.4419423 2.0531856 2.0531856 -106.97152
300 -104.78803 2.0263183 1.7037288 1.7037288 -106.81435
400 -105.12831 2.2305996 1.8754886 1.8754886 -107.35891
500 -105.38205 1.9777836 1.6629208 1.6629208 -107.35984
600 -105.59386 2.0222647 1.7003206 1.7003206 -107.61613
700 -105.7253 2.1621149 1.8179066 1.8179066 -107.88741
800 -105.68601 2.2922083 1.9272892 1.9272892 -107.97822
900 -105.45029 2.0621575 1.7338624 1.7338624 -107.51245
1000 -105.08573 2.0637772 1.7352243 1.7352243 -107.14951
1100 -104.62775 2.0877981 1.755421 1.755421 -106.71555
Actually, those output temperatures are pretty far away from what I set (300 K), so is there any notes I should care about when simulating ellipsoids and spherical hybrid systems? I noticed there are some questions and answers about this problem, but these do not match my question.
Have you noticed any warnings in the output? There should be some because you applying time integration to the same atoms multiple times. That is very bad.
Also, your setup is likely to trigger some limitations of the aspherical integrators when using fix npt/asphere, so I can only recommend against it unless you can use group all (which is impossible when you also have to use fix rigid or any of its variants)
Exactly, I fixed the problem by changing the command as:
group backbone type 1 2
group sidechain type 3
fix 33 sidechain npt/asphere temp 300 300 400 iso 1 1 4000
fix 11 backbone rigid/small molecule
Output: (T stablizes at around 300 K)
Step TotEng KinEng Temp c_myTemp PotEng
0 641.78583 335.44221 282.03987 282.03987 306.34362
100 638.45619 356.22374 299.51299 299.51299 282.23245
200 635.83582 351.68146 295.69384 295.69384 284.15436
300 629.66601 350.78763 294.94231 294.94231 278.87838
400 625.16026 320.77146 269.7047 269.7047 304.38881
500 620.74329 337.89465 284.10188 284.10188 282.84864
600 620.00748 322.66851 271.29974 271.29974 297.33897
700 622.08396 342.01434 287.56572 287.56572 280.06962
800 628.92604 358.95378 301.80841 301.80841 269.97226
900 643.16786 339.45742 285.41586 285.41586 303.71044
1000 650.08323 349.5181 293.87488 293.87488 300.56513
1100 651.62592 352.6732 296.52769 296.52769 298.95272
1200 651.44443 337.46307 283.73901 283.73901 313.98136
1300 649.69527 339.73751 285.65136 285.65136 309.95777
1400 647.07411 331.03525 278.33451 278.33451 316.03885
So as you answered, now I separately fix the backbone and sidechain using different fix commands, is it reasonable? Since the temperature is correct, I am not sure this treatment could be accepted? And I will test some physical properties of my polymer chain system. All in all, thanks for solving my questions, it really saves a lot of time.
It is required to have two integrators for this setup.
It looks like the temperature is a bit too low. Right now, you only thermalize the sidechains, but not the backbone. You can try using the “langevin” option to fix rigid/small to also thermalize the backbone particles and no only rely on transmission of kinetic energy (which should be decent, though, due to the construction of your model).
Yes, I have smapled the temperature response. When I use the following command:
group backbone type 1 2
group sidechain type 3
fix 33 sidechain npt/asphere temp 300 300 400 iso 1 1 4000
fix 11 backbone rigid/small molecule
I get the T for a long time simulation, which is 297 ± 11 K, and there is only one single chain in my system, so I think it is reasonable? I’ll check it by switching to a larger system later.
-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.
But when I use this following command combined with langevin:
group backbone type 1 2
group sidechain type 3
fix 33 sidechain npt/asphere temp 300 300 400 iso 1 1 4000
fix 11 backbone rigid/small molecule langevin 300 300 400 5451564
The average temperature outputted is 281 ± 11 K, which is a little bit lower than the value I set (300 K).
So I think maybe the former one is appropriate for me to carry out the simulation work? I am not very sure if there is some fatal error in this setting.
I cannot provide more help at this point because the choice of which settings you trust is ultimately yours. You cannot write in a paper “I just did what this dude on the internet told me” ![]()