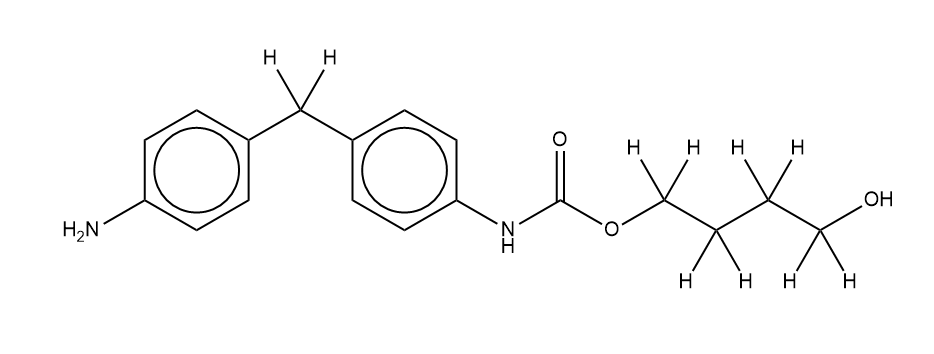
Dear,
when I tried to build molecule like as below with PCFF, EMC gives tips :
Warning: increment pair {o_2, c-} set to zero.
Warning: increment pair {c-, na} set to zero.
Warning: no torsion coefficients found for {hc, c, o_2, c-}.
Warning: no torsion coefficients found for {c, c, o_2, c-}.
Thank you for reporting the above issue. The problem stems from incorrect typing of the carbamate carbonyl carbon. This can be remedied by adding the following to your pcff_templates.dat file: