Dear Lammps users/developers,
I am encountering a problem with correctly defining orientation of ellipsoid particles in a coarse-grained (CG) model of benzene with each molecule is an ellipsoid.
Basically what I am trying to do is to output a CG ellipsoidal representation of my atomistic model of benzene molecules. To do this I calculated the principal axes and the quaternions from the atomistic configuration (using the moment of inertia to calculate the principal axes of each molecule, and calculate the quaternions from the rotational matrix whose column are the principal axis vectors arranged in a correct way to give det(matrix)=1, just in case if you wonder how I did this)
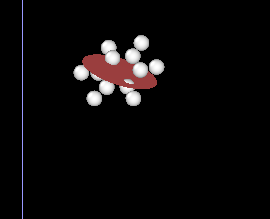
Then I wrote a Lammps data file for these ellipsoids using the calculated centre of mass, shapes and quaternions. I ran this model for 0 timestep to create a dump file with outputted quaternions (which are the same with what they are in the data file), and visualise this model with the atomistic model in OVITO. For some reasons the orientation of the particles in the two models do not agree with each other, the ellipsoids are oriented differently to the atomistic benzene molecules (but not perpendicular to each other, so this is not due to wrong settings of the ellipsoid shape parameters).
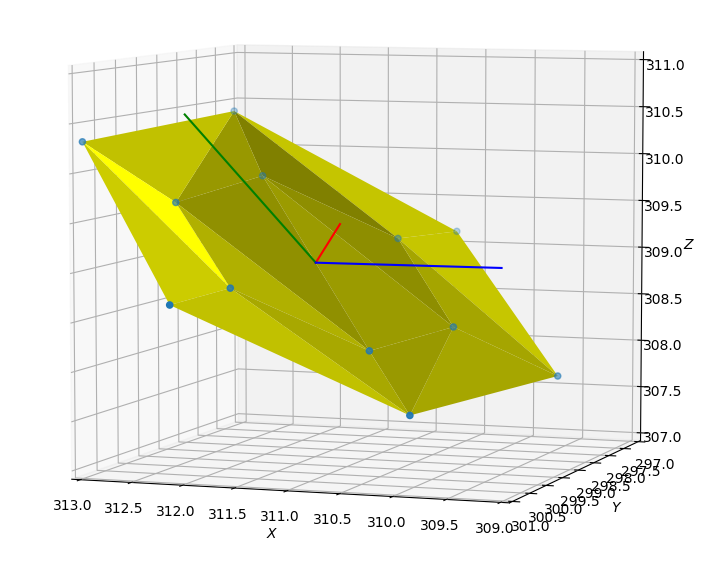
I know there is a description of quaternions in the Lammps manual, and I calculated quaternions using exactly the same methods (using the invariant rotational axis and angle). I am pretty sure my principal axes are correct, as I plotted these axes and the atom coordinates out using some codes, and they look just right (aligning with the molecular plane).
I don’t really understand why there is is mismatching going on. Any thought about this is much appreciated.
Ps: The lammps version I am using is 11Aug17. And I attach here 2 figures of the same benzene molecule: (1) plot of benzene molecular plane and calculated principal axes, and (2) atomistic model and ellipsoidal model in OVITO
Thank you,
Huong Nguyen
Dear Lammps users/developers,
I am encountering a problem with correctly defining orientation of ellipsoid particles in a coarse-grained (CG) model of benzene with each molecule is an ellipsoid.
Basically what I am trying to do is to output a CG ellipsoidal representation of my atomistic model of benzene molecules. To do this I calculated the principal axes and the quaternions from the atomistic configuration (using the moment of inertia to calculate the principal axes of each molecule, and calculate the quaternions from the rotational matrix whose column are the principal axis vectors arranged in a correct way to give det(matrix)=1, just in case if you wonder how I did this)
Then I wrote a Lammps data file for these ellipsoids using the calculated centre of mass, shapes and quaternions. I ran this model for 0 timestep to create a dump file with outputted quaternions (which are the same with what they are in the data file), and visualise this model with the atomistic model in OVITO. For some reasons the orientation of the particles in the two models do not agree with each other, the ellipsoids are oriented differently to the atomistic benzene molecules (but not perpendicular to each other, so this is not due to wrong settings of the ellipsoid shape parameters).
do i understand correctly, that with “run 0” you get the exact same output when comparing the files but different visualizations? that would make it not a LAMMPS problem, but a visualization program problem. have you contacted the OVITO author on this?
axel.
Dear Axel,
You’re correct. With run 0 Lammps does output exactly the same quaternions in the dump file.
This is indeed a mistake from me, I did not read the OVITO manual carefully. In OVITO the order of quaternions is (i,j,k,w) instead of (w,i,j,k) by default, so one needs to change these accordingly. When I changed to the correct order everything looks correct now.
Thanks a lot for helping me to realise my silly mistake.
Huong
Hi again,
Sorry but I have another question. How does LAMMPS defines the shape parameters of an ellipsoid with respect to its orientation?
I can see from ovito that some molecules fit with their ellipsoid model, but some of them are perpendicular to their ellipsoid model. I simply define the shape parameters in the same for all ellipsoids for now (because I’m not sure how to put them in shape_x shape_y shape_z correctly)
Huong
if you have a different question, then please post it as a new question with a suitable subject line.
otherwise people will likely think this to be a continuation of the discussion. this would be a problem, since all i can tell you is, what LAMMPS does is written in the documentation.
axel.