Comment: But if you are getting θ=179.99, then I suspect you are using a bond-angle force
with an equilibrium bond angle θ0=180 degrees. If that is the case, then there is no barrier
preventing θ->180. In that case, it simply does not make sense to apply a dihedral angle force
to this polymer. I suspect you might need to increase the complexity of the representation
of the polymer. A simple chain of beads is often not a good model of a polymer which can
support twisting. I suggest reading the simulation papers comming from
Andrzej Stasiak’s lab (Try reading the appendix from this paper.)
Yes, that’s true! Two of my angular potential have an equilibrium at 180. However, the exact
same simulation (with tabulated potentials) was done in GROMACS before.
There, it worked perfectly.
It sounds as if the GROMACS implementation of your model did not have this problem, but the LAMMPS conversion does. In that case, you probably made a mistake when you converted it into LAMMPS format. Mistakes like that are really easy to do.
All I can say is if any of the 4-body dihedral angle (φ) interactions in the model contain 3 atoms which (θ) approach 180°, then you are guaranteed to eventually have a numerical explosion in your simulation, regardless of the timestep. (Incidentally, 1fs is not that small, at least for all-atom simulations.)
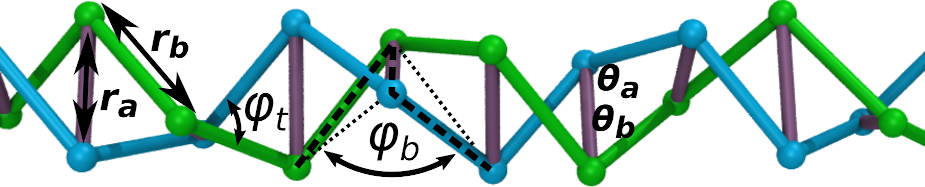
(It is possible to have a twistable polymer containing 180° bond-angles. As an example, the coarse-grained twistable polymer model that I mentioned earlier from Andrzej Stasiak’s lab has some 3-body bond-angles constrained to 180°, however those 3 atoms do not appear in any of the dihedral interactions. [2 of the 3 atoms do, but not all 3.] This is difficult to explain without drawing pictures, but I’m too lazy to do that. See the appendix of that paper.)
I had the task to export the Simulation to LAMMPS to check if
we could get a simulation speed up.
Compared to Gromacs? Seems unlikely. (I don’t know. LAMMPS has other nice qualities: flexibility, generalizability, numerical accuracy perhaps.)
First, I copied the tabulated potentials from GROMACS into LAMMPS, but with
LAMMPS the simulations used to crash after some time. Then, I changed from
tabulated potentials to LAMMPS potentials, but that did not help.
Does someone have an idea, why this problem is not showing up with GROMACS?
Perhaps when you converted the model into LAMMPS format, some of your angle or dihedral interactions are connected to the wrong atoms. Again this kind of mistake is easy to do. (Also check the two ends of the polymer to see if anything weird is happening there.)
I suggest building a small version of your system.
Try a polymer containing only 2-4 monomers and containing the minimal number of dihedral interactions (hopefully only 1).
See if the problem persists. If not, then keep adding monomers until the problem returns.
In addition, I would draw a cartoon stick-figure of your short polymer on paper and see where all of the 3-body angle and 4-body dihedral interactions are located. Make sure that none of the 3-body angle interactions with 180 equilibrium values contain 3 atoms which are participating in the same 4-body dihedral angle.
If the GROMACS model (or the script to convert it into LAMMPS) was written by somebody else, then for the sake of your own understanding, perhaps you should try building the system from scratch for LAMMPS instead of relying on somebody else’s work. (Assuming you are not already doing so.) It should not be that hard. The LAMMPS DATA file format is relatively easy to understand. You can write a script to create these kinds of files relatively easily.
Alternatively, you can try using one of the LAMMPS molecule-builders to do this for you. The topotools molecule builder might be useful to you, if you like VMD and don’t mind using TCL. I will also include a shameless plug for my own tool which I wrote (when I was in your position) to build general coarse-grained molecules and polymers in LAMMPS. (Examples: 1, 2, 3.) If your polymer is complex enough, or if you eventually plan to make the system complicated and add other molecules, then in the long run I suspect it might be less cumbersome to use this tool than to write your own script. You can use these instructions (1, 2, 3) to make the polymer wrap to any shape you desire (and then add additional bonds or branches in moltemplate if you need them). You can use moltemplate without sacrificing any understanding about what your system is or how it behaves. You’re still in control. It is not a black box tool. (Neither is topotools.)
(It actually seems that https://www.sciencedirect.com/science/article/pii/S0032386119304707?dgcid=rss_sd_all (Kubo et al., 2019),
had the same problem using LAMMPS)
If so, it is not because of a bug in LAMMPS.
Good luck!
Andrew
{kind=link}
{kind=link}
{kind=link}