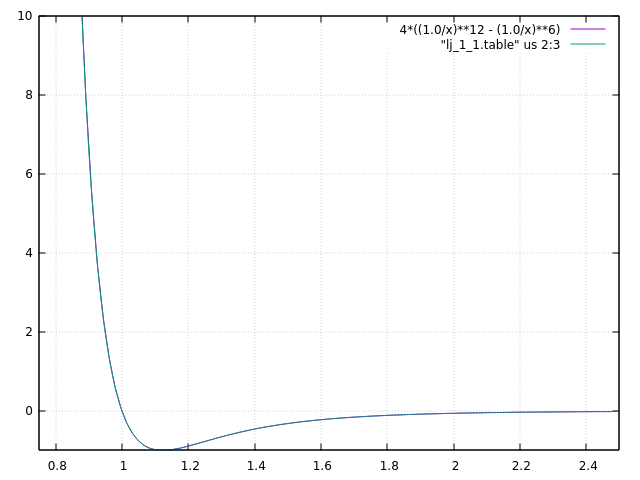
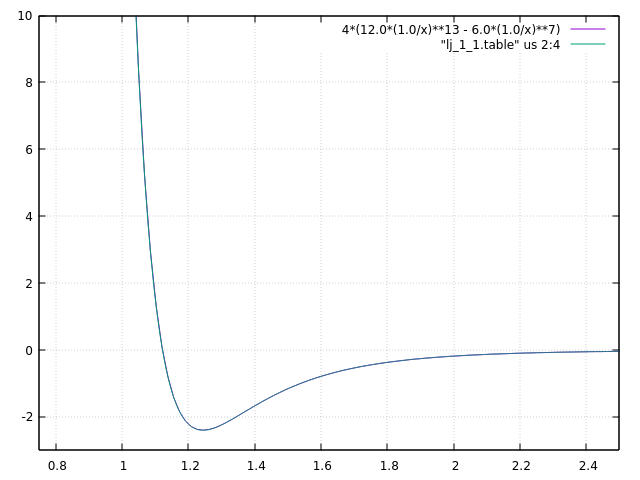
Dear all,
I need to use a pair_style hybrid/overlay including a pair_style table to implement a 12-6-4 Lennard Jones potential.
During the energy minimization I receive a lot of WARNING messages which state “WARNING: 1490 of 1801 force values in table are inconsistent with -dE/dr”.
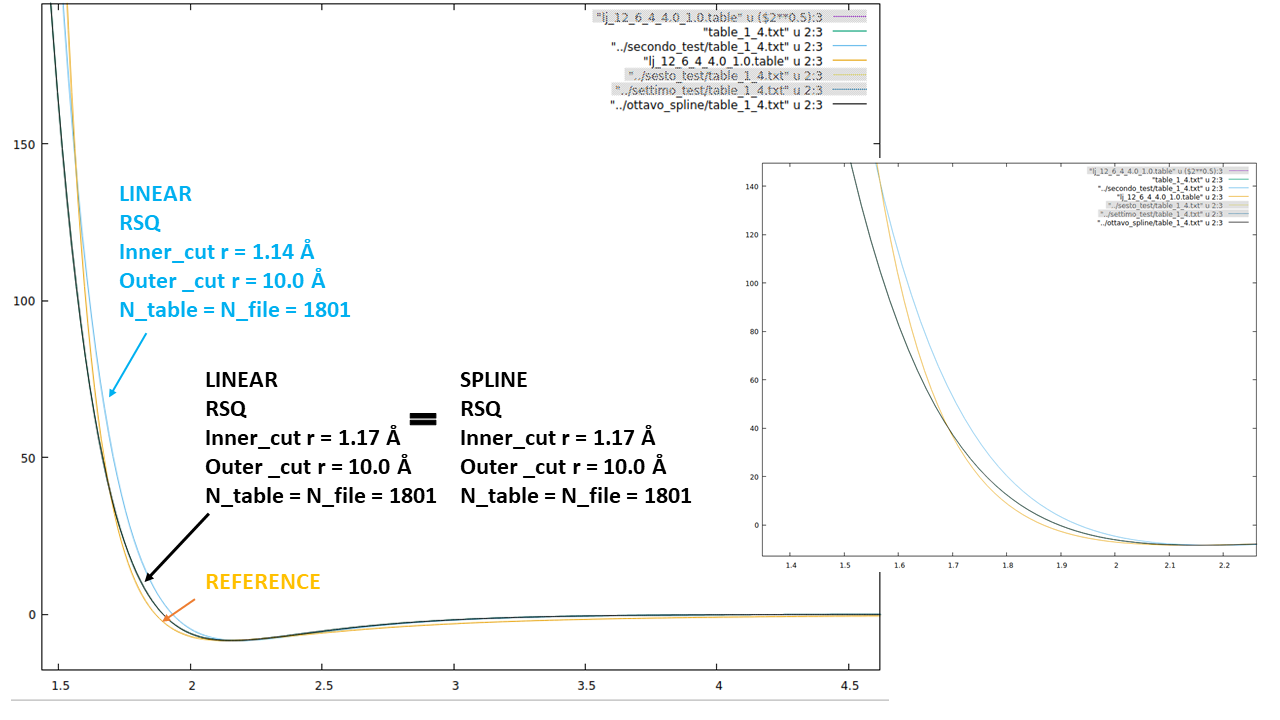
I tried to print the preliminary spline interpolation elaborated by LAMMPS, using the linear style, Ntable = Nfile and I tried both the R and the RSQ style. In my tabulated potential I considered the potential and the force at all distances between 1.0 Å and 10.0 Å to avoid fitting in the steepest region of the LJ repulsive branch and to use the same outer cutoff as the one used for the other pair_style respectively. I attach the lines of the test using RSQ style just as an example:
pair_style hybrid/overlay lj/cut/coul/long 10.0 10.0 coul/long 10.0 table linear 1801 ewald
pair_modify mix geometric
pair_modify tail yes
special_bonds lj/coul 0.0 0.0 0.5
kspace_style ewald 0.0001
include “pair_coeff.txt”
where pair_coeff.txt includes lines such as
pair_coeff 1 4 coul/long
pair_coeff 1 4 table lj_12_6_4.table POTENTIAL__1_4
pair_write 1 4 1801 rsq 1.0 10.0 table_1_4.txt POTENTIAL__1_4
The POTENTIAL__1_4 section of lj_12_6_4.table (my tabulated LJ) is the following
# DATE: 2022-25-10 UNITS: real CONTRIBUTOR: Emma (header line)
#12_6_4 potential for Zn (one or more comment or blank lines)
POTENTIAL__1_4
N 1801 RSQ 1.0 10.0
1 1.00 41531.50 503487.00
2 1.06 30056.62 355471.42
3 1.11 22103.39 255441.13
4 1.17 16490.52 186509.33
[…]
118 7.44 -4.38 -4.08
119 7.49 -4.32 -4.05
120 7.55 -4.26 -4.01
[…]
1800 99.95 -0.03 -0.01
1801 100.01 -0.03 -0.01
Increasing the distance, the potential has a minimum as expected from a LJ potential.
However, the file generated by LAMMPS is
# Pair potential hybrid/overlay for atom types 1 4: i,r,energy,force
POTENTIAL__1_4
N 1801 RSQ 1 10
1 1 83774.7220705805 1006396.11241192
2 1.02713192920871 60784.833626375 710594.257489771
3 1.05356537528527 44842.4797601862 510701.361776132
4 1.07935165724615 33584.5343687146 372962.001222688
[…]
118 2.7267196408872 76.1345668269373 68.8347968944961
119 2.7367864366808 75.4641403223875 68.1541852804469
120 2.74681633896407 74.8034404977079 67.4954486143175
[…]
1800 9.99724962177098 0.00431673939449543 0.0255875076741771
1801 10 0 0
I plotted the two potentials they look completely different (as it emerges from the values in the tables). The most worrying difference is that the LAMMPS potential has no minimum and never goes to negative values. As I said, I also tried using R instead of RSQ, I reduced the points (951), but LAMMPS generates exacly the same curve. I attach a graph.
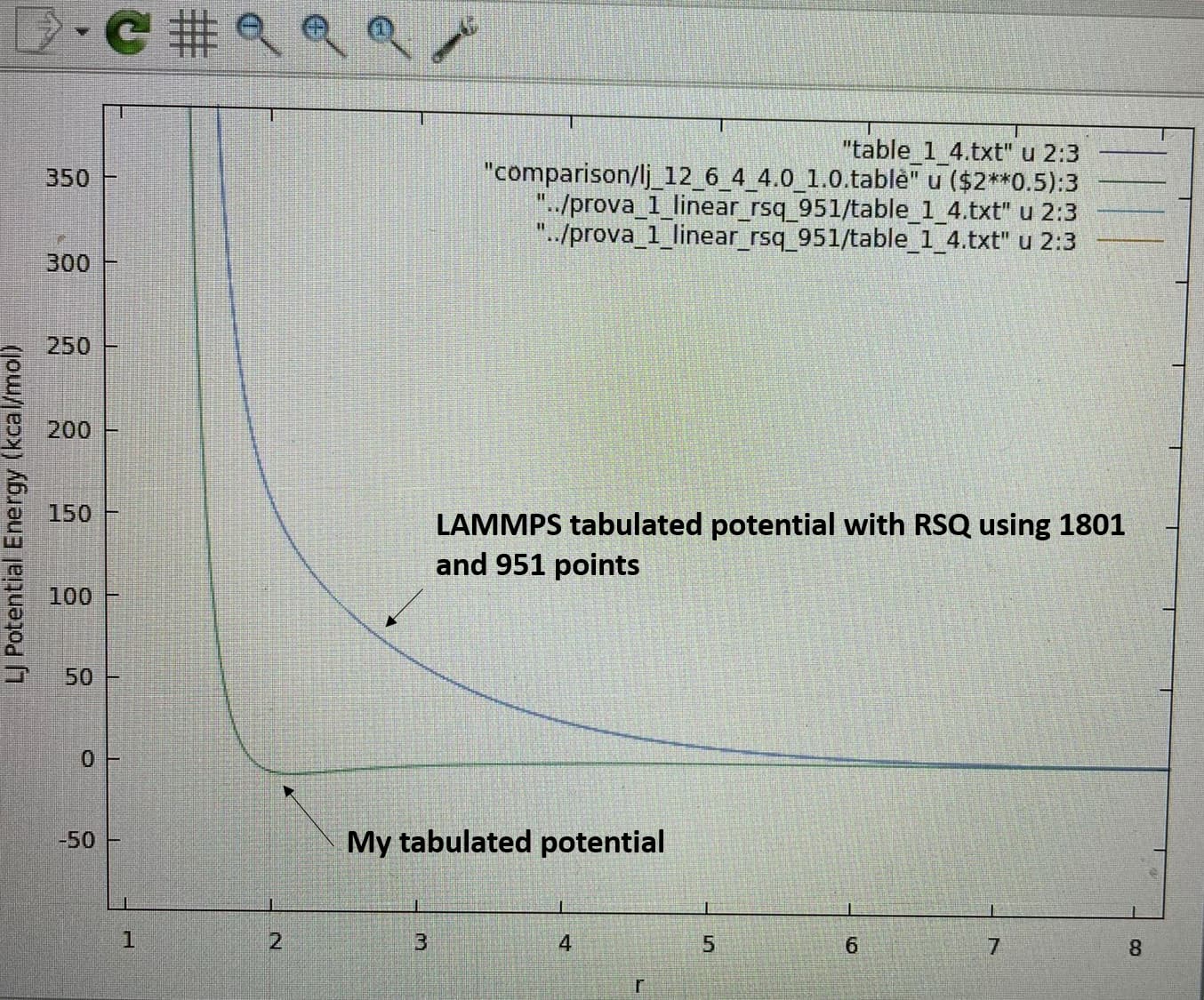
I cannot figure out what is the error I made in my input files since the RSQ of my table and the R of the LAMMPS table are consistent. I would be enormously grateful if someone could help me.
Thank you very much in advance for your kind collaboration.
Best regards,
Emma Rossi