Dear lamps-users,
I’ve established the SiC structures and first used NPT ensemble to equilibrium the whole system.
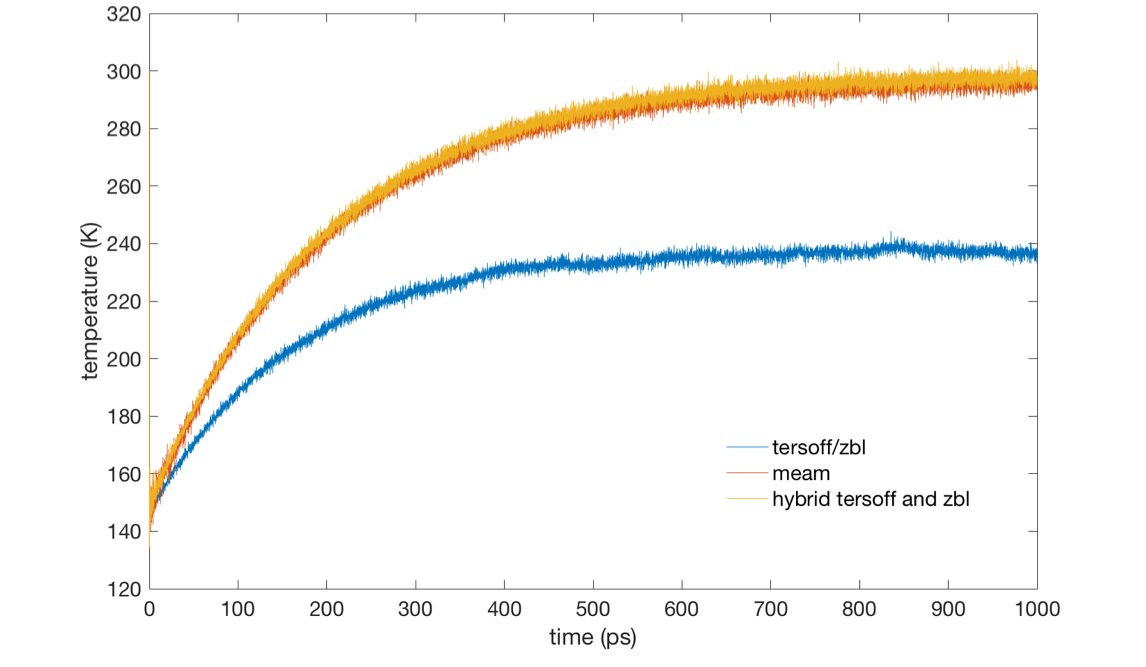
When I used the SiC.tersoff.zbl potential (obtained from LAMMPS library), the equilibrium temperature is 240K but I set this to be 300 K. And my input script is (there are only two types of atoms in the input data, C and Si):
pair_style tersoff/zbl
pair_coeff * * SiC.tersoff.zbl C Si
Then, I’ve tried the team potential (obtained from LAMMPS library) and to hybrid tersoff potential (obtained from LAMMPS library) and zbl potential, with which I both got the equilibrium temperature 300K. The other settings are with no difference.
pair_style meam
pair_coeff * * library.meam Si C SiC.meam Si C
pair_style hybrid/overlay tersoff zbl 3.0 4.0
pair_coeff * * tersoff SiC.tersoff C Si
pair_coeff 1 1 zbl 6.0
Pair-coeff 2 2 zbl 14.0
The temperature evolution with these three potentials is attached.
I’m confused if there’s some wrong with the input script using SiC.tersoff.zbl potential?
Thank you.
Best,
Wanzhen