Dear LAMMPS community,
I am conducting simulations to investigate phase transitions in metals. I have set up a simple bulk solid of aluminum, initialized at 400 K, then heated to 1325 K and cooled back to 400 K using a temperature ramp. However, I am encountering some problems related to the setup and the simulation. The script used is as follows:
clear
units metal
dimension 3
atom_style atomic
atom_modify map array
variable tempstart equal 1325
variable tempstop equal 425
variable myseed equal 12345
variable atomrate equal 1000
variable time_step equal 0.005
variable time_eq equal 100000
variable Vol equal vol
variable Temp equal temp
variable Press equal press
variable TotEng equal etotal
variable tdamp equal "v_time_step*1000"
variable pdamp equal "v_time_step*1000"
#------------
lattice fcc 4.048914 origin 0.0 0.0 0.0 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
region box block 0 4 0 4 0 4 units lattice
create_box 1 box
create_atoms 1 box
pair_style meam
pair_coeff * * library.meam Al Al.meam Al
#------------
reset_timestep 0
timestep 0.003
velocity all create 400 ${myseed} mom yes rot no dist gaussian
fix equilibration all npt temp 400 400 ${tdamp} aniso 1 1 ${pdamp} drag 0.2
fix 3 all print 1000 "${Temp} ${Vol} ${Press} ${TotEng}" append vol-temp-up-down2-1000-aniso-1000-4ec-eq.txt title ""
#fix 3 all print 1000 "${Temp} ${Vol} ${Press}" append vol-temp.txt title ""
thermo 1000
thermo_style custom step pxx pyy pzz lx ly lz temp etotal vol
dump dp0 all atom 100 dump-up-down2-1000-aniso-1000-4ec-eq.lammpstrj
run 50000
undump dp0
unfix equilibration
reset_timestep 0
fix melting all npt temp 400 1325 ${tdamp} aniso 1 1 ${pdamp} drag 0.2
fix 3 all print 1000 "${Temp} ${Vol} ${Press} ${TotEng}" append vol-temp-up-down2-1000-aniso-1000-4ec-take2.txt title ""
thermo 100
thermo_style custom step pxx pyy pzz lx ly lz temp etotal vol
#dump 1 all image fcc.Al.*.cfg mass type xs ys zs id
dump dp1 all atom 100 dump-up-down2-1000-aniso-1000-4ec-take2.lammpstrj
run 167000
unfix melting
fix cooling all npt temp 1325 400 ${tdamp} aniso 1 1 ${pdamp} drag 0.2
#fix 3 all print 1000 "${Temp} ${Vol} ${Press} ${TotEng}" append vol-temp-up-down2-1000-aniso-1000-4ec.txt title ""
thermo 100
thermo_style custom step pxx pyy pzz lx ly lz temp etotal vol
#dump 1 all image fcc.Al.*.cfg mass type xs ys zs id
#dump dp2 all atom 100 dump-up-down2-1000-aniso-1000-4ec.lammpstrj
run 167000
unfix cooling
1.) The output seems to be qualitatively acceptable, but it shows some artifacts. The expected result is a hysteresis in the temperature–energy graph, due to overheating and overcooling. However, the output I obtain is the following:
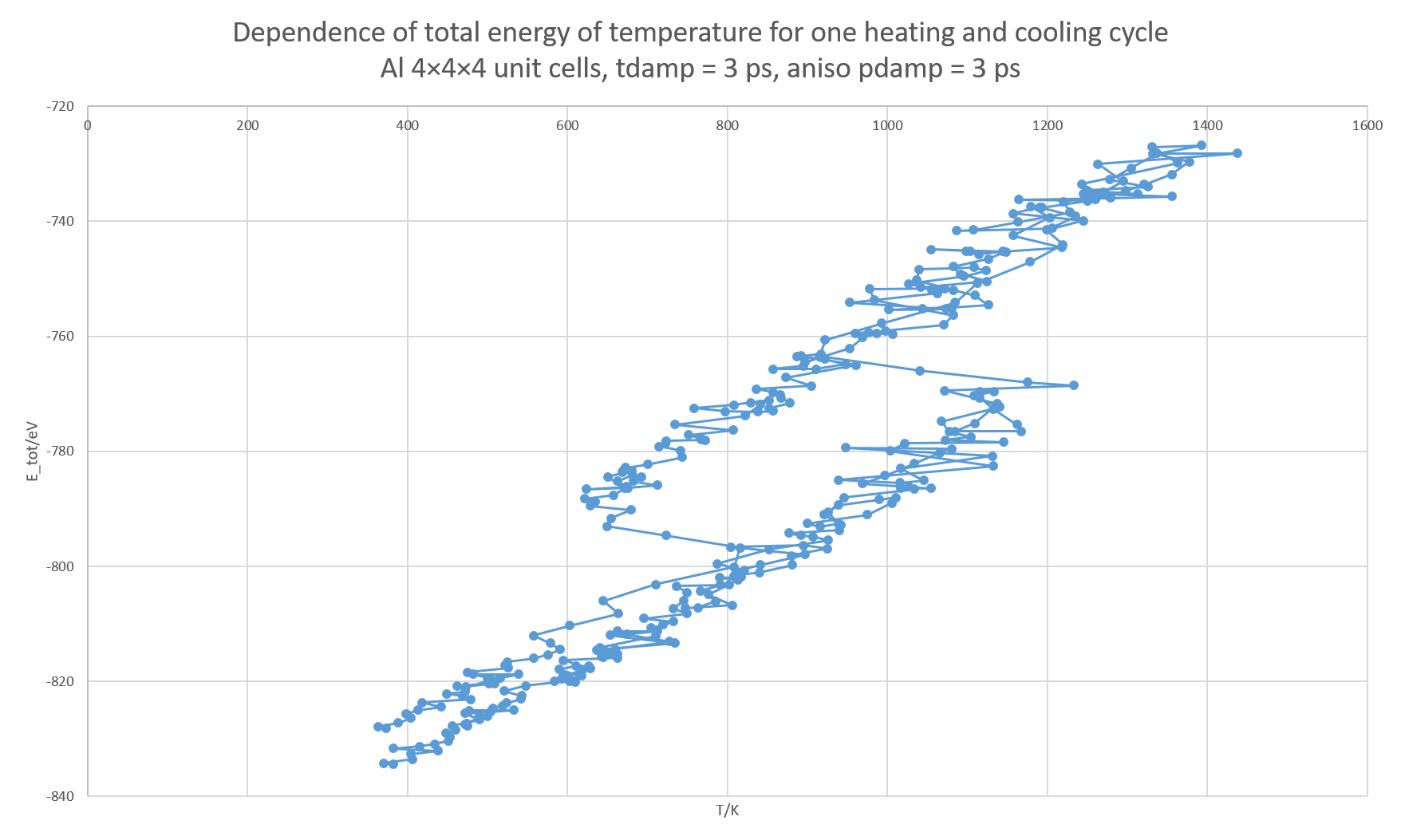
The expected behavior during a phase transition is that (e.g. during melting), the energy increases while the temperature remains constant. Here, the temperature drops rapidly while the energy mildly increases, followed by a quick increase in temperature and energy back to the temperature at which the melting started.

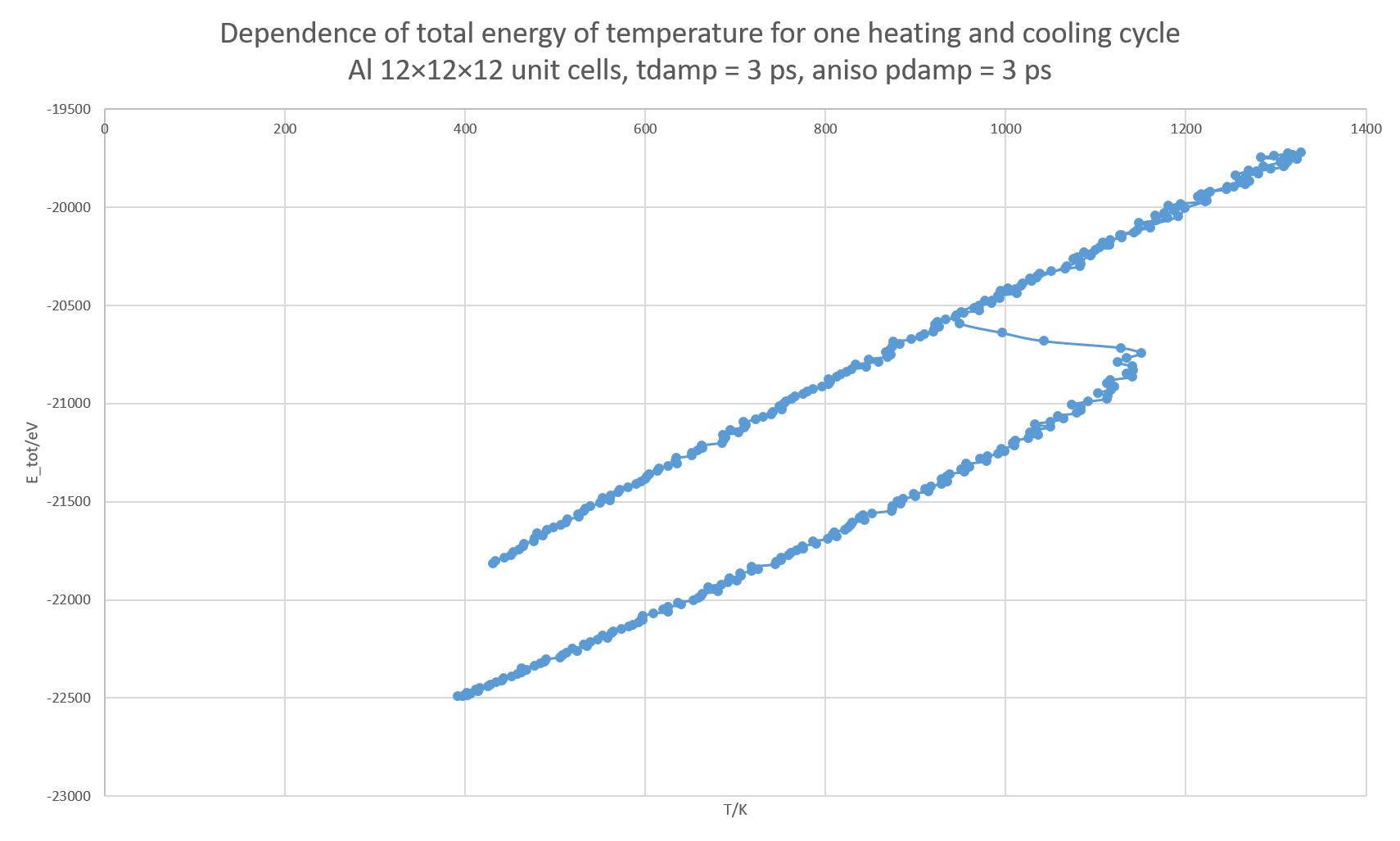
2.) If I increase the number of cells to 12^3, the undesired oscillations improve:
clear
units metal
dimension 3
atom_style atomic
atom_modify map array
variable tempstart equal 1325
variable tempstop equal 425
variable myseed equal 12345
variable atomrate equal 1000
variable time_step equal 0.005
variable time_eq equal 100000
variable Vol equal vol
variable Temp equal temp
variable Press equal press
variable TotEng equal etotal
variable tdamp equal "v_time_step*1000"
variable pdamp equal "v_time_step*1000"
#------------
lattice fcc 4.048914 origin 0.0 0.0 0.0 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
region box block 0 12 0 12 0 12 units lattice
create_box 1 box
create_atoms 1 box
pair_style meam
pair_coeff * * library.meam Al Al.meam Al
#------------
reset_timestep 0
timestep 0.003
velocity all create 400 ${myseed} mom yes rot no dist gaussian
fix equilibration all npt temp 400 400 ${tdamp} aniso 1 1 ${pdamp} drag 0.2
fix 3 all print 1000 "${Temp} ${Vol} ${Press} ${TotEng}" append vol-temp-up-down2-1000-aniso-1000-eq.txt title ""
#fix 3 all print 1000 "${Temp} ${Vol} ${Press}" append vol-temp.txt title ""
thermo 1000
thermo_style custom step pxx pyy pzz lx ly lz temp etotal vol
dump dp0 all atom 100 dump-up-down2-1000-aniso-1000-eq.lammpstrj
run 50000
undump dp0
unfix equilibration
reset_timestep 0
fix melting all npt temp 400 1325 ${tdamp} aniso 1 1 ${pdamp} drag 0.2
fix 3 all print 1000 "${Temp} ${Vol} ${Press} ${TotEng}" append vol-temp-up-down2-1000-aniso-1000-take2.txt title ""
thermo 100
thermo_style custom step pxx pyy pzz lx ly lz temp etotal vol
#dump 1 all image fcc.Al.*.cfg mass type xs ys zs id
dump dp1 all atom 100 dump-up-down2-1000-aniso-1000-take2.lammpstrj
run 167000
unfix melting
fix cooling all npt temp 1325 400 ${tdamp} aniso 1 1 ${pdamp} drag 0.2
#fix 3 all print 1000 "${Temp} ${Vol} ${Press} ${TotEng}" append vol-temp-up-down2-1000-aniso-1000.txt title ""
thermo 100
thermo_style custom step pxx pyy pzz lx ly lz temp etotal vol
#dump 1 all image fcc.Al.*.cfg mass type xs ys zs id
dump dp2 all atom 100 dump-up-down2-1000-aniso-1000-take2.lammpstrj
run 167000
unfix cooling
but the issue mentioned in point 1 remain. Worse still, the system appears amorphous /liquid already at 400 K. Also, the hysteresis observed with 4^3 cells disappears, as the system no longer crystallizes during cooling.
In summary: What causes this unphysical setting at the outset of the simulation and at the phase transition points? Could it be due to inadequate values of tdamp and pdamp (thermostat/barostat damping parameters) ?
Thanking you in advance, I look forward to your advice.