Hi all,
is there any chance to continue the computation of with respect to the initial coordinates?
I’ve just run a couple of tests and found that using the outdated compute msd/molecule and running from a restart-file, the msd is computed with respect to the current coordinates in the restart-file. Here’s the essential part of my script:
read_restart diff.800.restart
#read_data init.data
…
thermo_style custom step temp vol etotal pe ke epair ebond eangle edihed press density
thermo 10
timestep 1.0
label diff
restart 100 diff.*.restart
compute 2 all msd/molecule
fix 1 all nve
fix 2 all langevin 300.0 300.0 100.0 12345
fix 3 all ave/time 1 100 100 c_2 file msd.dat mode vector
run 3000000
unfix 4
unfix 3
unfix 2
unfix 1
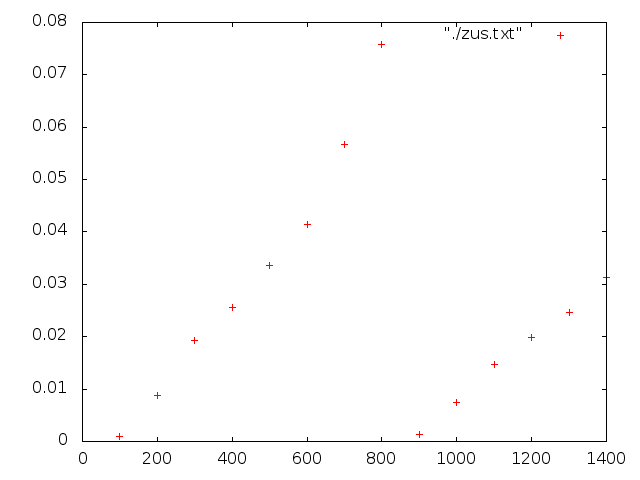
The 2nd run i restarted from step 800, i used the same ID for my computes and still, the compute is re-initialized…see the appended png (msd vs step)
any hints? Maybe i should use compute cc1 all chunk/atom molecule and compute msd all msd/chunk cc1 ?
thx in advance,
regards,
frank.