I am trying to execute a lammps script on reverse osmosis, when I run the calculation, it’s blocked in the first step of the calculation, as it reached the running step, I supposed that I didn’t have errors in the script, the picture below is the message it’s blocked at.
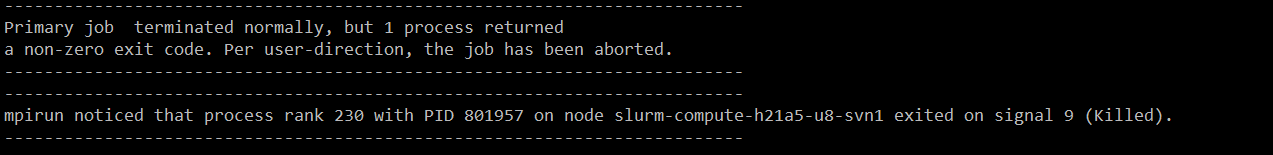
After almost 10 min I receive the following message.
The message is from the MPI library and indicates that one of your parallel processes was killed by the batch system. This says nothing about what happened. The most common reason is that your simulation had some bad geometry with close contacts causing one atom to move very far within a single timestep, so that it cannot be wrapped back into the principle simulation cell, but without having access to the console output of the simulation, there is nothing to confirm what this was about.
Thank you for your feedback, I didn’t want to reply till I give some trial and forwarded you with my reflections
1-I tried to review my geometry created in Packmol, defined by a tolerance of 2.0 A between every two atoms. I increased it to 3.0 but I received the same error.
2-Concerning the potential I chose values from the literature for Lennard-jones potential that worked for previous simulations. Do you think this can be the cause?
3- In my simulation too, I have periodicity in the three directions, so I am wondering if lammps consider the atoms in the edges too close to the new atoms generated by periodicity.
WARNING: Proc sub-domain size < neighbor skin, could lead to lost atoms (src/domain.cpp:936)
You are running a system with only 2653 atoms on 224 MPI processes. That is extremely wasteful and likely much slower than running with much fewer processes since with such small subdomains, most work will be needed to update the ghost atom information. As a rule of thumb, most systems do not show much performance improvements with fewer than 1000 atoms per processor.
The fact that the simulation reaches this point only confirms that there are no syntax errors in the input, it can still be (scientifically) bogus and have bogus energies/forces. You won’t see much of that when running in parallel, so before even running in parallel, I would test this on a desktop machine.
Also, you need to do some scaling benchmarks to see how much speedup you can get by running in parallel. I would be surprised if you can retain parallel efficiency beyond 10 MPI processes.
Hello Axel, I would like to thank you for your previous feedbacks.
A new update concerning my reverse osmosis simulation progress:
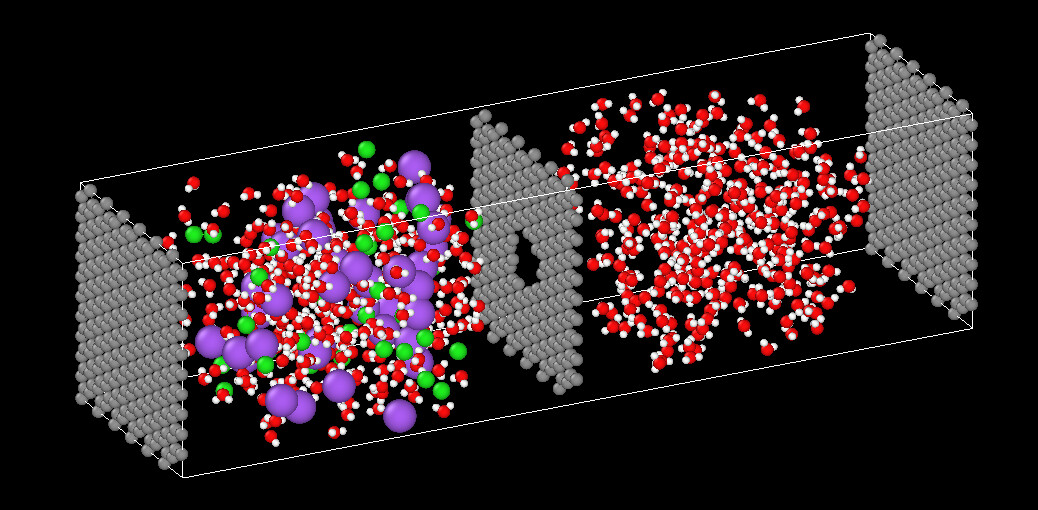
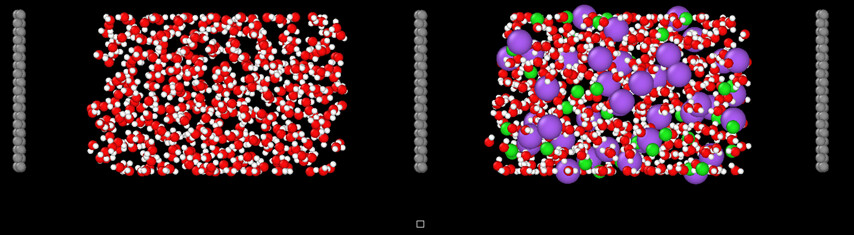
I discovered I had an issue with my simulation cell coordinates 'generated by VMD) which were too small compared to my geometry these two figures show how I increased it.
In fact, the message error is about:
Out of range atoms - cannot compute PPPM (src/KSPACE/pppm_tip4p.cpp:106)
It’s been several days since I tried these paths to resolve the issue:
1-Increase the number of atoms but I received the same error type
2-decrease the times step from 0.5 to 0.1 but nothing changed
3-I increase the neighboring frequency changing into this command neigh_modify delay 0 every 1
But I received a new error about bond atoms missing as described in this output file. slurm-1566763.out (15.4 KB)
I would appreciate receiving your opinion about my simulation, and if anyone else had the same issue please send me your feedback.
I am suspicious about using AIREBO to model a single layer graphene sheet. In your situation, I would start with simple Lennard-Jones particles for the carbon and have them immobile, before trying something like AIREBO. I also think you need a slab correction instead of full periodicity but I doubt that would crash your simulation.
Then again, I am a stranger on the Internet and unfamiliar with AIREBO (sadly I am often suspicious of unfamiliar things). If you know for sure that AIREBO routinely produces sensible results in the same situation you’re deploying it, feel free to let me know I am wrong.
Also, why not write_data after your minimised system and see what has happened during minimisation?
I cannot really give any significant comments because most of the important information is not shown.
The kind of error you observe is quite common for systems with a bad geometry or a bad choice of force field (or force field parameters).
Also, using 224 processors on such a tiny system with less than 5000 atoms is a massive waste. I would be surprised if this system scales to more than 10 processors. At the same time, with this size you will not get meaningful results either due to finite size effects.
But this all has very little to do with LAMMPS and a lot with understanding, planning, and debugging simulations and making important choices for your research, which makes it mostly off-topic for this forum and a topic to discuss with your adviser/tutor.