Dear LAMMPS users,
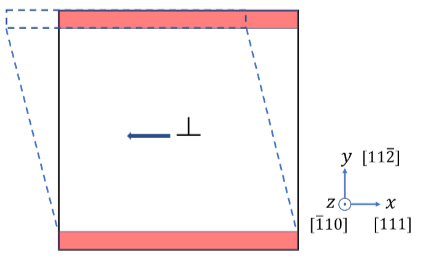
I am working on calculating the Peierls stress of an edge dislocation in an HCP crystal using molecular statics simulations in LAMMPS. My goal is to obtain the (overall) stress associated with the dislocation motion. I introduce an edge dislocation (generated using Atomsk) into a simulation box of size Lx × Ly × Lz = 100b × 100b × b, where b is the lattice constant. The boundary conditions are: periodic in the x and z directions and shrink-wrapped in the y direction. The dislocation line is aligned along the z direction, while its glide direction is along x. I limit the simulation box in the z direction to a single lattice period (1b) to prevent the distortion of the dislocation line during loading. To apply shear loading, I define upper and lower atom groups and iteratively displace the upper group in the x-direction, keeping the lower part fixed (see the image). The relevant LAMMPS script snippet is:
label loop
displace_atoms lower move ${dx_per_step} 0.0 0.0 units box
min_style cg
minimize 1e-14 1e-14 10000 10000
next a
jump SELF loop
To compute the shear stress, I sum up the xy component of the per-atom stress as follows:
compute stress_mob mobile stress/atom NULL
compute stress_mob_red mobile reduce sum c_stress_mob[4]
Then, I divide this quantity by the simulation volume (Lx × Ly × Lz) to obtain the shear stress. However, I see two peculiar behaviors in the shear stress-shear strain response: 1) large oscillations, and 2) as I increase the simulation box size, I expect the shear stress to eventually converge to a well-defined value. However, instead of stabilizing, the response keeps growing (see the image below). This is unexpected, as I assumed that beyond a certain system size, size effects should diminish, leading to a converged solution.
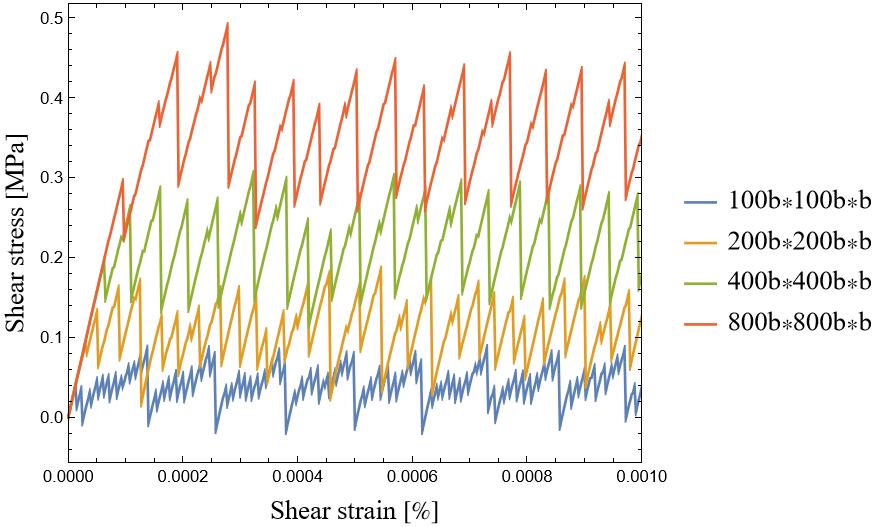
So, here are my question:
- Am I correctly calculating the shear stress?
- Is there a better approach for simulating this molecular statics scenario?
Any insights, suggestions, or relevant references would be greatly appreciated.