Hello, I am trying to calculate the average volume of my box during a NPT calculation, and compare the result from the output data file to be sure that I get what I want, but these two do not agree.
I use a fix all ave/time command with the same Nevery, Nrepeat, and Nfreq both to print the out on the file.dat and to calculate the average volume inside Lammps. The time is resetted to zero before the definition of the two fixes.
Actually I think the results should be the same independently of the specific N chosen. I tried with the options ave one, ave window 1 or just none, and it seems the result isn’t the same.
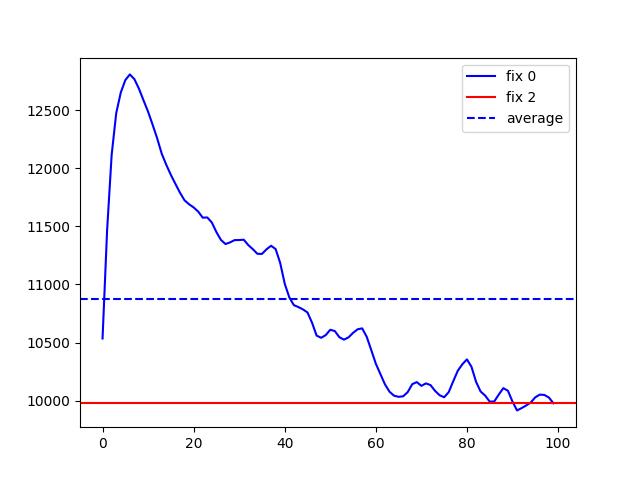
For more context, I want to calculate the average volume in NPT so that I can rescale the box and fix the volume in NVT to calculate GK viscosity. If I use the one printed by the fix, the cell looks too big (and the average pressure slightly negative).
Hi Marco,
I can’t comment on the use of a variable to call a fix ave/time (f_2), as I never use it this way. I can however confirm that measuring the average value from f_0 as you do is a valid approach, as long as you disregard the beginning of the simulation for which the volume did not converge to its equilibrium value…
Simon
edit: After looking closer at your input, the value of ${mean_Vol} from the print command makes sense, the variable mean_Vol is called at the very end of the simulation, so it returns its last value that was obtained by averaging over the last 100 steps.
There is not enough information here to make a meaningful assessment.
For example, have you established that the system is already fully equilibrated?
how large is the system and how large the volume fluctuations and what is the typical frequency of those?
Are you sure that 10000 MD steps is a long enough simulation to get a proper average? Specifically for volume fluctuations the characteristic times can be rather long. It is much easier to expand a dense system than to condense it (again).
Thank you for your answers. Indeed in the real calculation the volume is averaged after equilibration is met. The system contains 1039 atoms and the input files were generated with packmol/moltemplate with input files from the ATB.
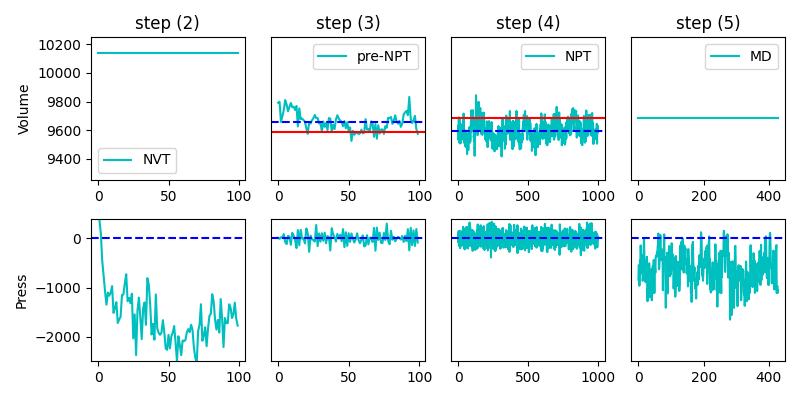
The full simulation includes (1) energy minimisation, (2) NVT equilibration, (3) pre-NPT equilibration to reach the equilibrium volume, (4) NPT equilibration and finally (5) MD run (NVT to calculate GK). The time step is 1fs.
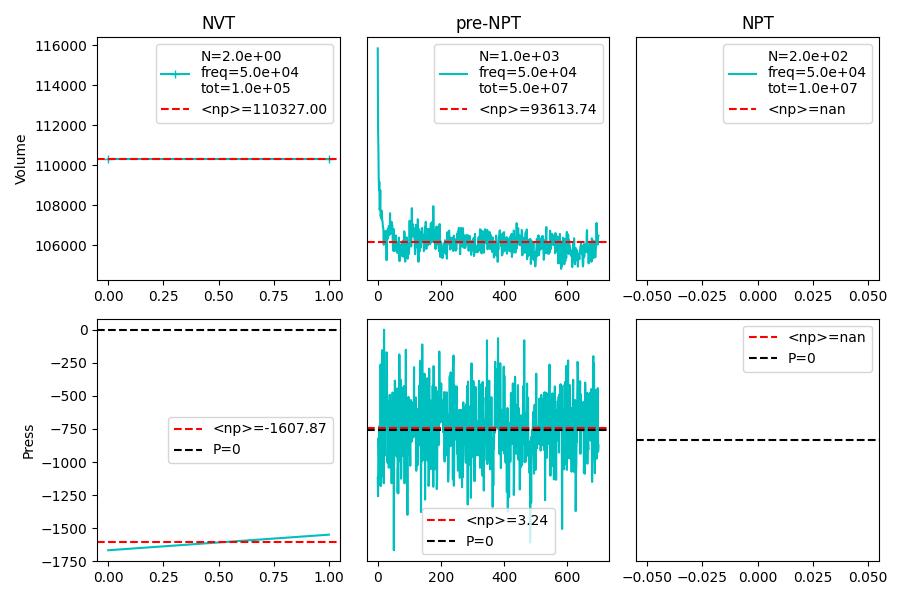
Please find below the full calculations. Note that I rescale the last volume before fixing NVT in (5) in order to meet the average volume obtained in step (4). In the figure, the average of the volume (blue) does not correspond to the output of the fix 2. Thus, the volume of the MD production run sits on the wrong value.
@simongravelle if you have other suggestions to calculate the average I am happy to consider them. I imagined defining a volume counter to increment at every step and divide by the number of steps, but this is deprecated in a number of lammps posts.
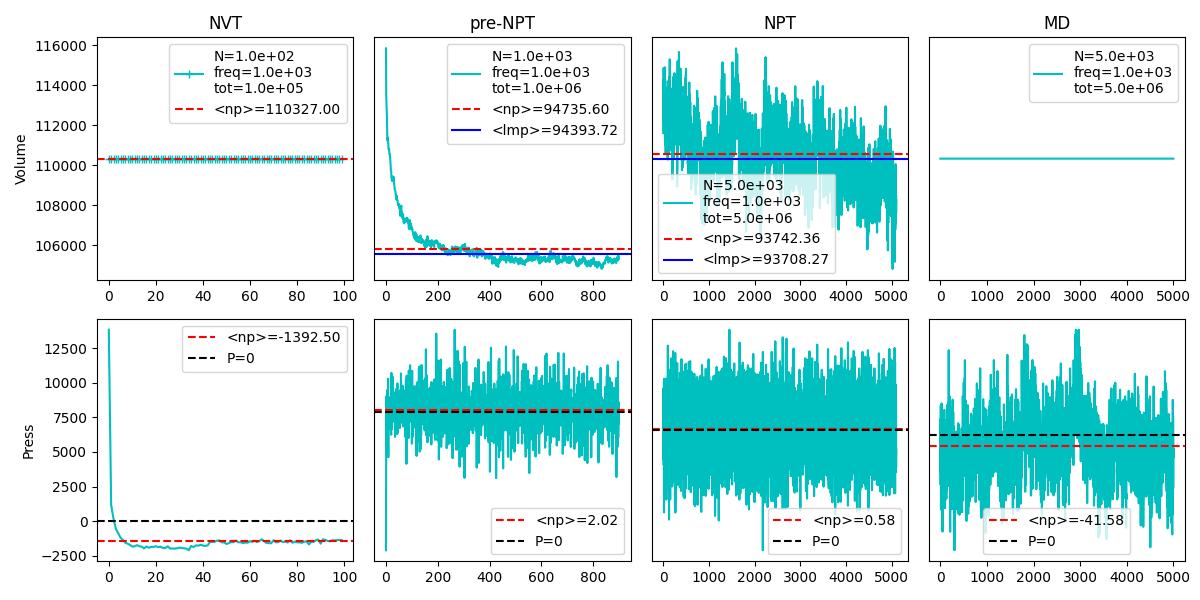
When I look at the data, it seems to me, that you need to do more equilibration and also need to average over more data to get a meaningful average of the volume. Only the second half of “step(4)” seems to be somewhat consistent (the first half may still have some memory of some initial setup non-equilibrium configuration) and in that second half you appear to have only about 5 periods of the characteristic vibration of the cell volume. That is clearly not enough to determine whether you have reached and maintained equilibrium. Of course you would need to average over the entire step(4) data starting from 500 time units to get a reliable average.
What is making the situation much more difficult is the extremely small size of the system with only about 1000 atoms. This will lead to rather large pressure fluctuations, and thus larger volume fluctuations and there are likely finite size issue. Thus for reliable statistics and data you would not only need to run for a longer time, but also over a significantly larger system (easily 10x larger, but 100x larger would not be excessive either).
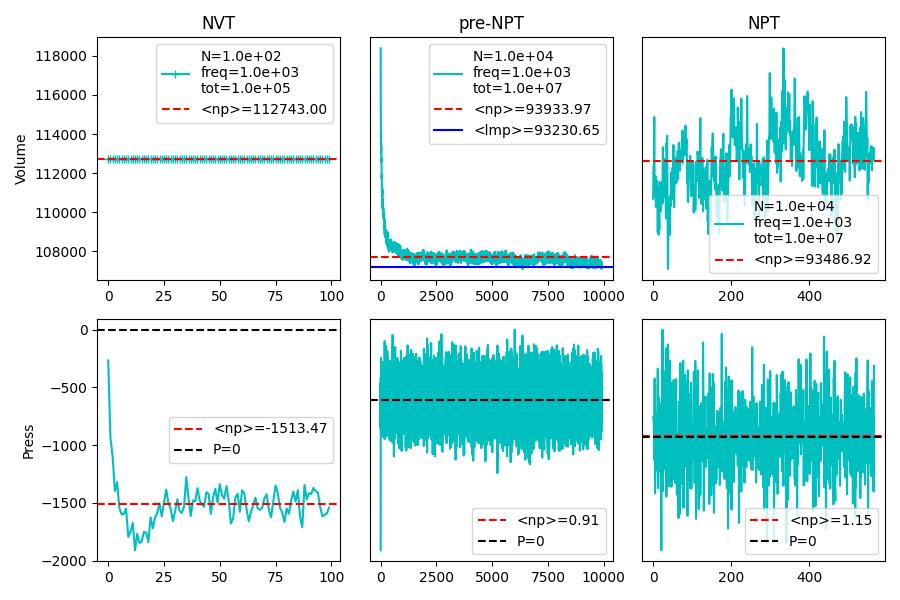
Thank you for your suggestions. I tested the same structure with 10k atoms and with larger time - and it’s clearer that after 5M timesteps of 1fs the average volume is still decreasing. The numpy average of the pressure values in the MD phase is still 40 atm far off.
I increased the NPT pre equilibration to a total of 1e7 time steps, and I get an average pressure around 1 atm - I guess I can take this as a good result?
I stopped this calculation and resent it with a lower output frequency and waiting for its results (it is still in the pre-NVT phase), although having less points make the average probably worse.
However, my initial question was about the correctness of the analytical derivation. Shouldn’t the average calculated by the fix 2 be analytically the same as the average of the values printed out in equilibration.dat no matter the number of points/state of the equilibration?
It is not easily possible to figure out what you are comparing to what here since your input file is complex and riddled with variables and thus extremely difficult to read and figure out from the outside.
You may notice that in every case you have investigated so far, the printed value of f_2 seems unrelated to the average of the f_0 printouts and suspiciously close to the very last value in the f_0 printout instead.
This may give you enough of a hint to attempt the scientific method. It always helps to have a short example to quickly iterate over hypotheses, so maybe you can experiment with a short run, like 100 steps?
Thank you @srtee, I believe you are right about try the method on a short run, and I have tried a very short run at the very beginning. What do you think is to be modified there?