Hello, user everyone
I have some questions about using API.
The question content is a bit long, but I appreciate your understanding and would be grateful if you could answer just once.
Detail Qeustions:
-
Is it correct that in the command
mpr.get_structure_by_material_id(material_id = MPID, final = True),final = Falserepresents the crystal structure provided by ICSD, whilefinal = Truerepresents the structure after VASP Relaxation? -
The web download structure seems to be conventional, and
mpr.get_structureappears to be a primitive structure (beforeget_primitive). How can I obtain the structure provided by the website? While using theto_conventional()option might be a way, this method has the drawback of potentially damaging the crystal structure by spglib or being unable to use the MAGMOM data stored in the site properties. -
In the case of
final = True, MAGMOM is stored in the site property column and printed together. Is this result the calculated magnetization, and does its order match the atomic order obtained through the structure? -
If the previous
structureis converted to a primitive cell throughget_primitive_structure(), can the order be considered properly maintained? While the visible order seems to match, I’m unsure if the order appropriately reflects properties like MAGMOM. -
Can Antiferromagnetic calculations be appropriately performed with the structure obtained from
get_primitive_structure()? To my knowledge, Antiferromagnetic calculations are precisely calculated using the conventional structure. -
What were the inputs for calculating the band and various data on the website, and where can I view the outputs of these results? Is the data obtained through
provenance_docs = mpr.materials.provenance.search(material_ids=[material_id])correct?
- Code -
for idx, structure_summary in enumerate(structures,1):
try:
# --- 1. 데이터 수집 ---
material_id = structure_summary.material_id # MPID
(...)
# --- 2. Get structure and preprocessing ---
structure = mpr.get_structure_by_material_id(material_id, final=use_initial_structure)
if isinstance(structure, list):
structure = structure[0]
(...)
primitive_cell = structure.get_primitive_structure()
(...)
else:
# 2) site_property mode: site_properties["magmom"] Used
if "magmom" not in primitive_cell.site_properties:
# Warning if no site_property exist
msg = f"[{material_id}] ERROR: site_properties['magmom'] Not exist!"
print(msg)
mag_info_for_log = "ERROR: no site_properties['magmom']"
The symmetry error while using primitive cell
By obtaining the structure through get_primitive_structure(), the following errors often occur. The reason is probably that there is a problem with the structure conversion processed by spglib.

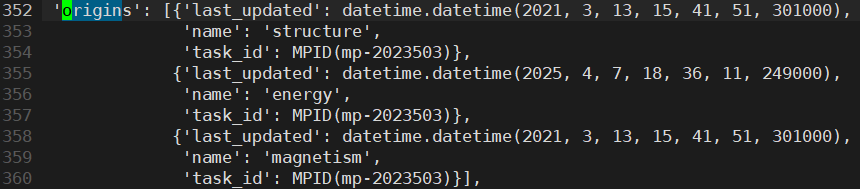
Most important question, what structures are the VASP calculation data provided by Materials Project based on?
The structures downloaded from the web seem to be provided based on conventional cell, but is there a way to see the structure actually used for writing material properties (band structure and magnetization, etc.) like the code where you could see the DOS structure?
Thanks for leading these long questions.
Best regards,
G. H.
- The code that build static calculation structure I use.
SCF_Structure_Builder.py (13.4 KB)