Hello,
I am trying to study the interaction of graphene with a polymer.
I used the Nanotube builder in VMD to build a graphene sheet. I have generated a LAMMPS data file for just the graphene sheet using VMD topotools. However, when I try to view the data file using readlammpsdata, and give "pbc box command, it shows the following:
ERROR) Suspicious pbc side length (a=65.769997 b=57.959999 c=0.000000). Have you forgotten to set the pbc parameters?
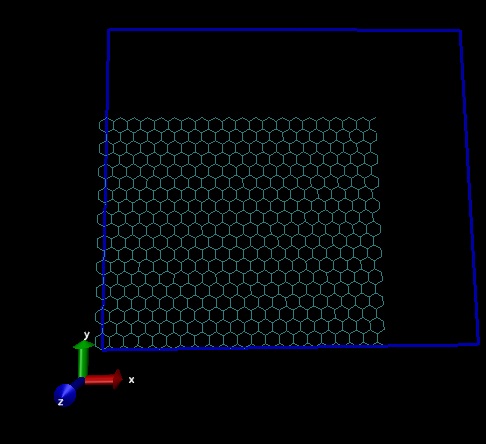
And irrespective of the xhi, xlo and yhi, ylo values in the data file (I have tried different sizes of the box), the box always is as shown in the picture attached.
When I run the LAMMPS script with this data file (a 2D simulation), it runs for a few steps and displays:
ERROR on proc 0: Bond atoms 587 588 missing on proc 0 at step 202 (…/neigh_bond.cpp:65)
I would like to know ways of rectifying this. My apologies if this is a bad query to ask.
Regards,
L Madhavkrishnan
BITS Pilani Hyderabad Campus
Hello,
I am trying to study the interaction of graphene with a polymer.
I used the Nanotube builder in VMD to build a graphene sheet. I have
generated a LAMMPS data file for just the graphene sheet using VMD
topotools. However, when I try to view the data file using readlammpsdata,
and give "pbc box command, it shows the following:
ERROR) Suspicious pbc side length (a=65.769997 b=57.959999 c=0.000000). Have
you forgotten to set the pbc parameters?
please note that this is a VMD question and thus should be directed to
the VMD mailing list.
VMD cannot always assumes that you provide a 3d input. you have to
manually override the size of the z dimension to be able to use
pbctools in VMD.
And irrespective of the xhi, xlo and yhi, ylo values in the data file (I
have tried different sizes of the box), the box always is as shown in the
picture attached.
same as above, this is a VMD question. you have to provide an example
file and state which VMD and topotools versions you are using.
When I run the LAMMPS script with this data file (a 2D simulation), it runs
for a few steps and displays:
ERROR on proc 0: Bond atoms 587 588 missing on proc 0 at step 202
(../neigh_bond.cpp:65)
this usually is an indication of a bad choice of potential parameters,
simulation parameters or a bad geometry input.
axel.