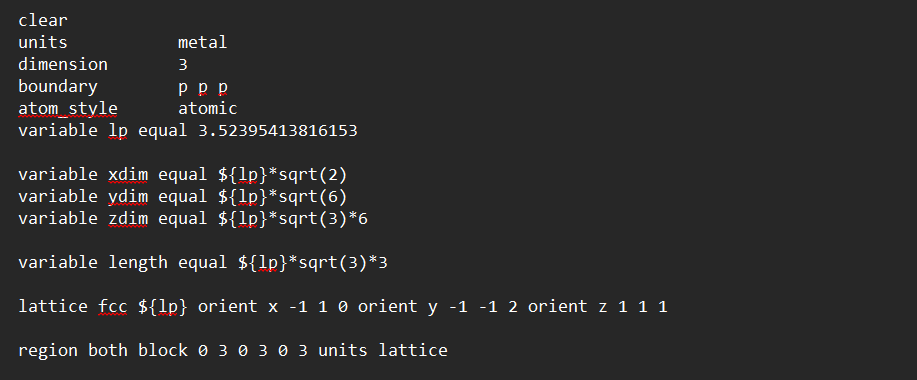
I am trying to create an FCC cell such that, z direction is 111 direction and x and y are perpendicular to them. check the following commands.
clear
units metal
dimension 3
boundary p p p
atom_style atomic
variable lp equal 3.52395413816153
variable xdim equal {lp}*sqrt(2)
variable ydim equal {lp}*sqrt(6)
variable zdim equal ${lp}*sqrt(3)*1
region test block 0 {xdim} 0 {ydim} 0 {zdim} units box
lattice fcc {lp} orient x -1 1 0 orient y -1 -1 2 orient z 1 1 1
create_box 1 test
create_atoms 1 region test
There are no issues till now, but i am trying to multiply x and y dimensions with 2 to double the system in respective directions. But on checking the structure in ovito it is not FCC, some other structure are also present.
why does it happen on doubling xdim and ydim but not when im replicating the box,
where is the periodicity broken?
dimensions are same as dir. magnitude
On doubling dimensions.
First off, do you think the subject line of your post describes what you are asking about?
If you want people to look at your problem, you need a descriptive subject. Yours says “I have a question”, which applies to almost every post and thus will for the most part indicate that you don’t know how to write a proper post and thus people will avoid even looking at it.
Please look at those. Can you read them well? There are detailed explanations in the forum guidelines post on how to correctly quote text from input files and similar in this forum.
This looks to me that this is a question you should ask in the OVITO forum elsewhere on the MatSci server. Visually, I don’t see a difference in the crystal structure in the two images. So why are you asking how LAMMPS creates an unwanted structure and not why OVITO is not detecting the structure as expected?
Why do you explicitly compute the lattice spacings and not let LAMMPS do that and use “units lattice” to define the box?
Why don’t you just use the replicate command?
Apologies for the way I wrote the post and LAMMPS commands. But I have tried creating region using units lattice, here is how the structure looked in OVITO. Here green ones are FCC and the rest are other type.
Here is the command:
I am fine with making structure with box, and open to any suggestions regarding that.
I basically want to create an intrinsic stacking fault in this. For that I have to displace the top region a/sqrt(6) in y axis, as y direction is the 112 direction.
Here is how I am displacing:
and here is the structure after displacement (green ones are FCC and white are of some random type (not BCC,HCP)
and here is the structure before any displacement (green ones are FCC again).
(ignore the dimensions of box, they are different from the ones mentioned in commands as I replicated in OVITO to visualize better).
My guess is I am doing wrong in magnitude of displacement. Am I right?
Thanks in Advance
You apologize, but you don’t correct your subject and malformatted post. Instead you are posting a new message with screenshots, which is as bad as the malformatted text, since one cannot cut-n-paste it to quickly test your code. This suggests that you did not spend any time reading the forum guidelines. You are certainly still not following them.
How do you expect people think of you if you ignore their suggestions like this?
So what? As I mentioned before, this looks more like an issue of OVITO than of LAMMPS, possible due to rounding. If you want to prove that LAMMPS is producing incorrect positions, you need to provide better and more convincing proof.
Mind you this part of the LAMMPS code has been around for a very long time, so it is highly unlikely that there is something wrong with LAMMPS. Tens of thousands of LAMMPS users before you would have noticed.
Science is not a guessing game. Whether the displacement is done the way that is correct for your research is not something that an outside can easily tell. You have to first and foremost discuss this with people that are familiar with and care about your research like you adviser or supervisor or tutor or experienced colleagues.