I have a problem with creating perfect crystals via LAMMPS.
For example, I want to create a single crystal Cu with the following (part of) commands:
# create geometry lattice fcc 3.6147 orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
region whole block 0 20 0 20 0 20 units lattice create_box 1 whole create_atoms 1 box
and compute the cna/atom with the command
#compute cna/atom compute cna all cna/atom 3.085334
I got a perfect crystal with all cna/atom equal to 1 (atoms of this kind are red). Then I change the orients to orient x -1 1 2 orient y 1 -1 1 orient z 1 1 0:
…
# create geometry lattice fcc 3.6147 orient x -1 1 2 orient y 1 -1 1 orient z 1 1 0
region whole block 0 20 0 20 0 20 units lattice create_box 1 whole create_atoms 1 box
…
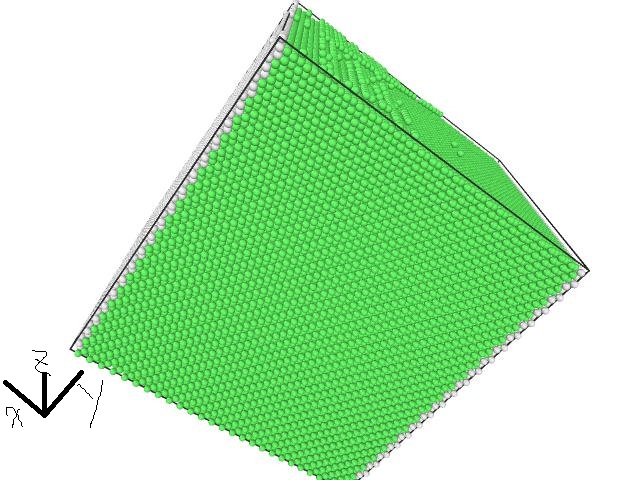
In this case ,the cna of the atoms at the boundaries along positive and negtive x-directions is 5 (atoms of this kind are gray). Also, the atoms at the boundaries along positive and negtive y-directions are not uniformly distributed. The snapshot is attached in this email.
Does anyone know what is the reason? How can I create a perfect crystal with custom orients?
I have a problem with creating perfect crystals via LAMMPS.
For example, I want to create a single crystal Cu with the following (part
of) commands:
# create geometry
lattice fcc 3.6147 orient x 1 0 0 orient y 0 1 0 orient z 0 0
1
region whole block 0 20 0 20 0 20 units lattice
create_box 1 whole
create_atoms 1 box
and compute the cna/atom with the command
compute cna/atom
compute cna all cna/atom 3.085334
I got a perfect crystal with all cna/atom equal to 1 (atoms of this kind are
red). Then I change the orients to orient x -1 1 2 orient y 1 -1 1 orient z
1 1 0:
...
# create geometry
lattice fcc 3.6147 orient x -1 1 2 orient y 1 -1 1 orient z 1
1 0
region whole block 0 20 0 20 0 20 units lattice
create_box 1 whole
create_atoms 1 box
...
In this case ,the cna of the atoms at the boundaries along positive and
negtive x-directions is 5 (atoms of this kind are gray). Also, the atoms at
the boundaries along positive and negtive y-directions are not uniformly
distributed. The snapshot is attached in this email.
Does anyone know what is the reason?
most likely you don't have the correct periodicity. i think with x -1
1 2 you need a box length divisible by 3. this has been discussed in
great detail in the past a few times. you should be able to find the
discussion in the mailing list archives.
You need to set the ‘spacing’ in each direction according to the length of the repeat vector. This is usually the vector described by the Miller indices.