I am trying to use the dump command to output the unwrapped atomic coordinates at each time step for use in postprocessing and have run into an intermittent and puzzling error. We see the problem with the 1Feb14 and 28Jun14 codes. The dump command is
dump 7 all custom 1 atomlist.txt xu yu zu
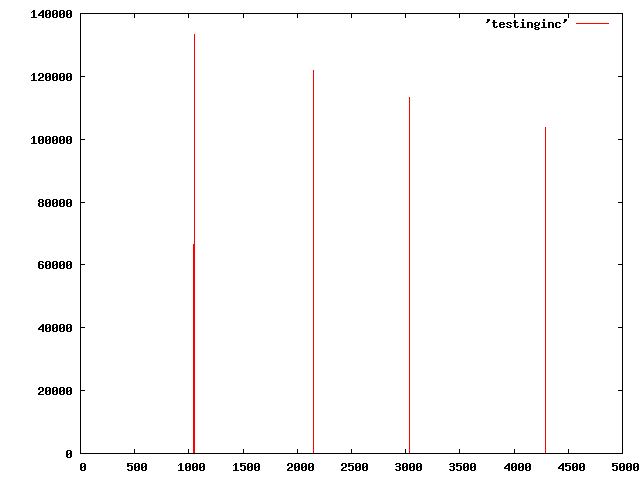
In our case, there are 685 atoms and the problem can be demonstrated for a few 1000 time steps. We wrote a program to read the atomlist.txt file and find that at random times, the incremental distance of the atoms at time t+dt relative to t is a very large number, while usually it is 0.0001-0.001. A plot of the distances squared per timestep is attached. I know it is my job to trace this error, but perhaps someone has a suggestion of whether the atom order is not being preserved or perhaps the unwrapping is incorrect or ???
The manual pages suggest that this should not happen for the custom dump, but perhaps I did not understand the fine print… Is there a better way to record the atomic positions than from this dump statement? Thanks in advance for your advice.
Natalie Holzwarth
N. A. W. Holzwarth email: natalie@…3310…
Department of Physics web: http://www.wfu.edu/~natalie
Wake Forest University phone: 1-336-758-5510
Winston-Salem, NC 27109 USA office: Rm. 300 Olin Physical Lab