Dear all
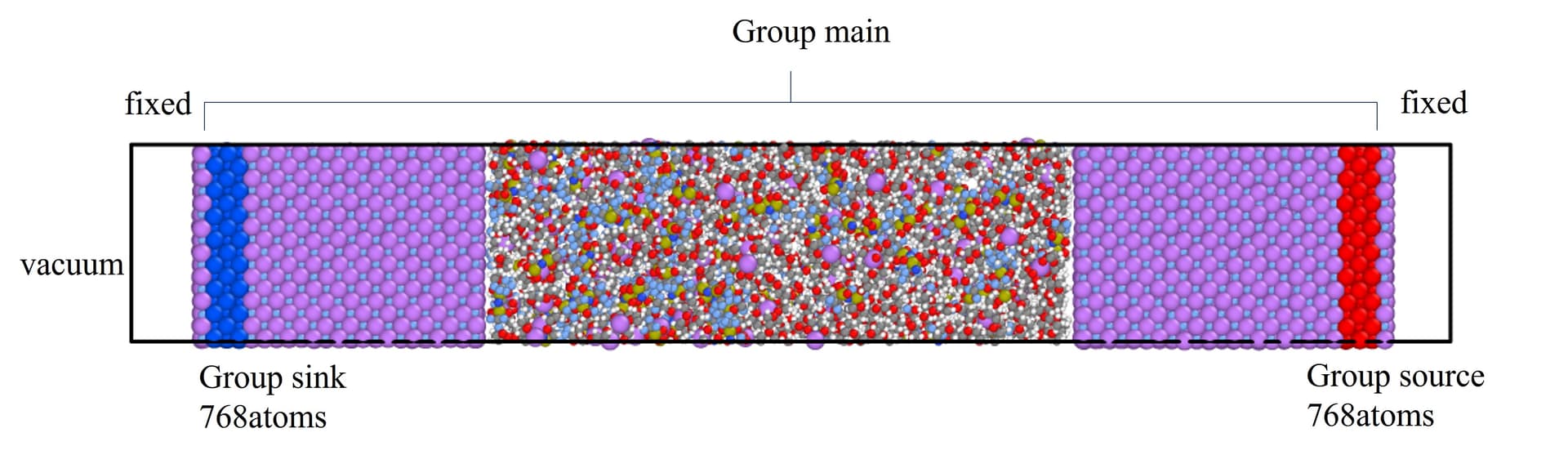
I’m trying to calculate the interfacial thermal conductivity between LiF and polymer(PEO) by NEMD method. The simulation model is showed as below. It contains group source, gourp sink, the main sample, two fixed layers, two vacuum layers at both ends. Source gourp and sink group have the same number of atoms which is 768.
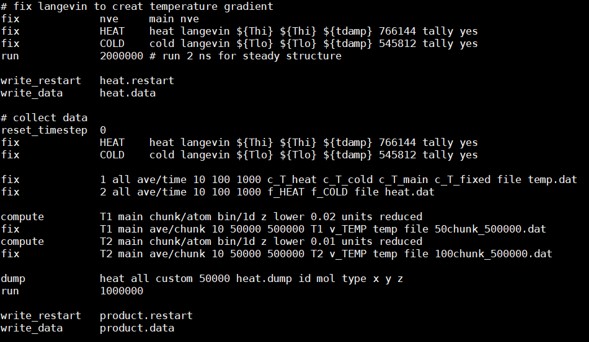
I controlled the heat source temperature to 355K and the heat sink to 255K by using fix langevin.The specific settings about this part is illustrated below. And before that, I have used the npt ensemble to stabilize the system at 305K. .
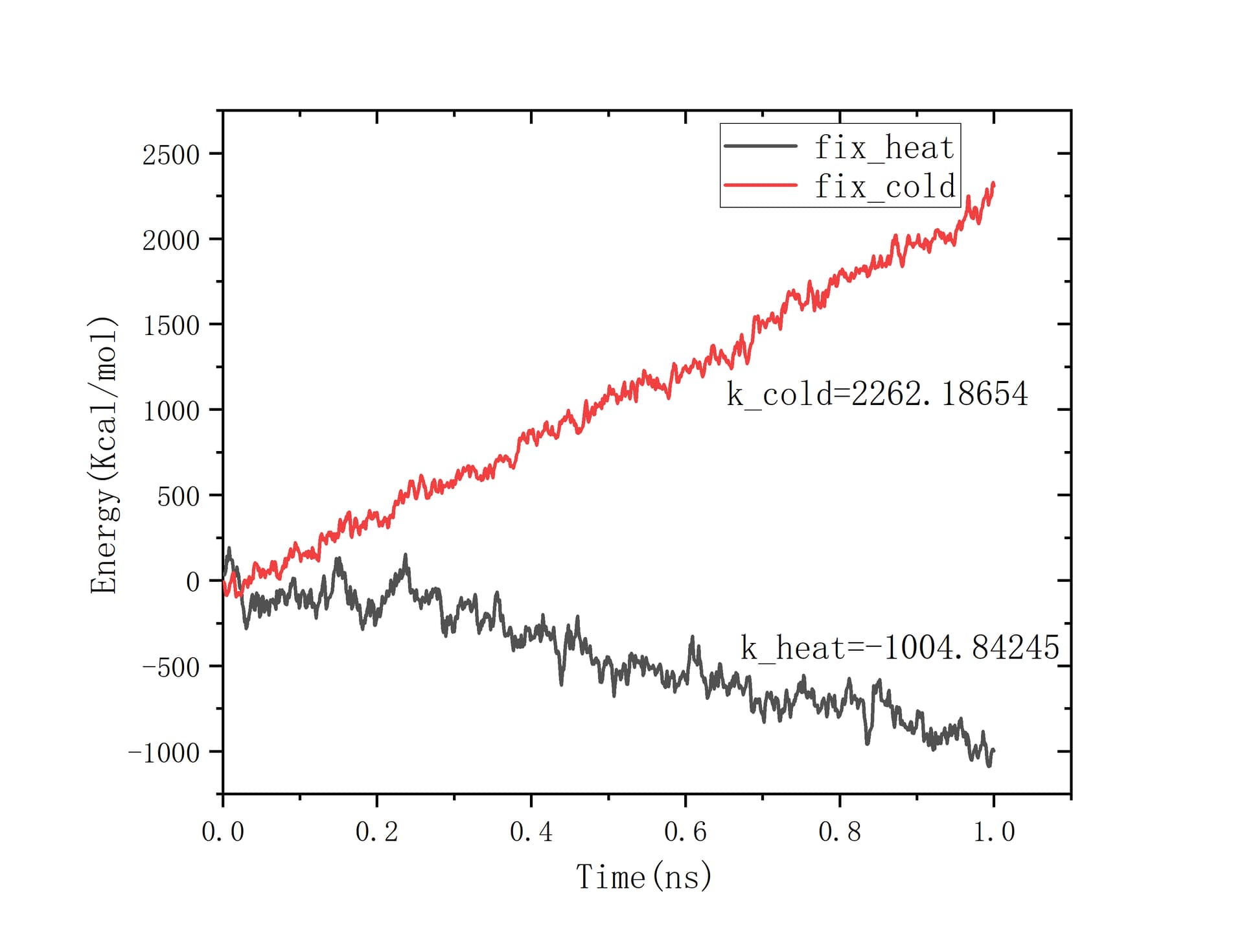
But when I tried to calculate the heat flux using the data producted from “fix heat” and “fix cold”, I found they differ from each other a lot. The slope of energy with respect to time is showed below.
In general, their gap should be small. So what’s the reason for that big difference between the energy accumulated in source and sink ?
Any comment on this issue is highly appreciated.
255 K is -20°C. My first guess would be that your cold region is freezing the surrounding PEO and that you are recording the continuous removal of enthalpy of freezing at that interface.
Notice how the heat outflow there begins immediately with a consistent gradient, whereas the heat inflow from hot region appears to only start with some lag. This may be consistent with an initial enthalpic “pulse” from the onset of freezing.
I don’t have any other guesses (and it IS a guess that you will have to assess further by yourself unless you provide further data).
This is likely not something that somebody can just tell from looking at your input (if it was that easy, we would not need to do simulations in the first place), but here are a couple of questions that may help you look for explanations.
Does your simulation conserve energy without the hot/cold region thermostats?
Is there a converged and linear temperature gradient across the system?
Thank you for your reply! I check the temperature of PEO and the lowest point of PEO temperature is about 236K. Is that mean PEO is frozen? And I don’t know much about the phenomenon of “enthalpic pulse” . But I will check it out. Thanks again!
It’s impossible for me to tell from here whether your PEO is frozen, or if that is even the source of the problem. Remember that the real-life freezing point of a substance is not guaranteed to be the freezing point that the force field exhibits in a simulation.
Simply downloading and visualising a trajectory will tell you whether the polymers are locally frozen or not.
“Enthalpic pulse” is a made-up phrase. It essentially means that if you had a polymer equilibrated at a temperature at which it is fluid, and it is suddenly cooled to freezing, the enthalpy of freezing must go somewhere. And if the cold source thermostat is already removing heat at its maximum rate, then the rest of the PEO must heat up while the steady state is being reached… And during that time the hot thermostat will have to add less heat to the system.
I am purely guessing the most interesting possible explanation. Whether it is valid or not, I am not even sure, and you will have to double check your own simulation results.
Dear Mr. Srtee:
I’m sorry for not getting back to you in a timely manner. Your idea is enlightening. I conducted the simulation again. In terms of the possible freezing phenomenon. I checked the trajectory as you said. It seems the polymer is not frozen. The gif below is a screenshot of the fix langevin process from 1.0ns to 2.0ns.
reset_timestep 0
timestep ${TS}
thermo 1000
thermo_style custom step temp vol press pe ke etotal
fix e_output all ave/time 100 10 1000 v_energy file e_output.dat
fix nvt all nvt temp 255 255 ${tdamp}
run 500000
unfix nvt
write_restart nvt.restart
write_data nvt.data
# define regions
variable z_l equal zlo
variable z_h equal zhi
variable z1 equal ${z_l}
variable z2 equal ${z_h}
variable c equal ${z1}+8
variable h equal ${z2}-8
variable fix_left equal ${z1}+2
variable fix_right equal ${z2}-2
region lfixed block INF INF INF INF ${z1} ${fix_left} units box
region rfixed block INF INF INF INF ${fix_right} ${z2} units box
region fixed union 2 lfixed rfixed
region cold block INF INF INF INF ${fix_left} ${c} units box
region heat block INF INF INF INF ${h} ${fix_right} units box
region main block INF INF INF INF ${fix_left} ${fix_right} units box
group fixed region fixed
group heat region heat
group cold region cold
group main region main
compute KE main ke/atom
variable KB equal 8.617333262145e-5*23.0609 # convert to kcal/mol
variable TEMP atom c_KE/${KB}/1.5
compute T_heat heat temp/region heat
compute T_cold cold temp/region cold
compute T_main main temp/region main
compute T_fixed fixed temp/region fixed
# fix boundary
velocity fixed set 0.0 0.0 0.0
fix fixed fixed nve/noforce
# add vaccum
change_box all z delta -10 10 units box
write_data vaccum.data
# fix nvt
fix nvt main nvt temp ${T} ${T} ${tdamp}
run 500000
unfix nvt
write_restart nvt_main.restart
write_data nvt_main.data
# fix langevin to creat temperature gradient
fix nve main nve
fix HEAT heat langevin ${Thi} ${Thi} ${tdamp} 766144 tally yes
fix COLD cold langevin ${Tlo} ${Tlo} ${tdamp} 545812 tally yes
run 2000000 # run 2 ns for steady structure
unfix e_output
write_restart heat.restart
write_data heat.data
# collect data
reset_timestep 0
fix HEAT heat langevin ${Thi} ${Thi} ${tdamp} 766144 tally yes
fix COLD cold langevin ${Tlo} ${Tlo} ${tdamp} 545812 tally yes
fix 1 all ave/time 10 100 1000 c_T_heat c_T_cold c_T_main c_T_fixed file temp.dat
fix 2 all ave/time 10 100 1000 f_HEAT f_COLD file heat.dat
compute T1 main chunk/atom bin/1d z lower 0.02 units reduced
fix T1 main ave/chunk 10 50000 500000 T1 v_TEMP temp file 50chunk_500000.dat
compute T2 main chunk/atom bin/1d z lower 0.01 units reduced
fix T2 main ave/chunk 10 50000 500000 T2 v_TEMP temp file 100chunk_500000.dat
dump heat all custom 50000 heat.dump id mol type x y z
run 1000000
write_restart product.restart
write_data product.data
During 1.0ns and 3.0ns, I used langevin to first get a stable structure before I collect the energy of sink and source. But I still get the same results as yesterday. Therefore, How do I determine the heat flow through the interface if the heat sink and the heat source energy are still not conserved?
Thanks for providing more information. The script portion you have pasted is still missing important data (such as the values of the timestep and equilibration temperature).
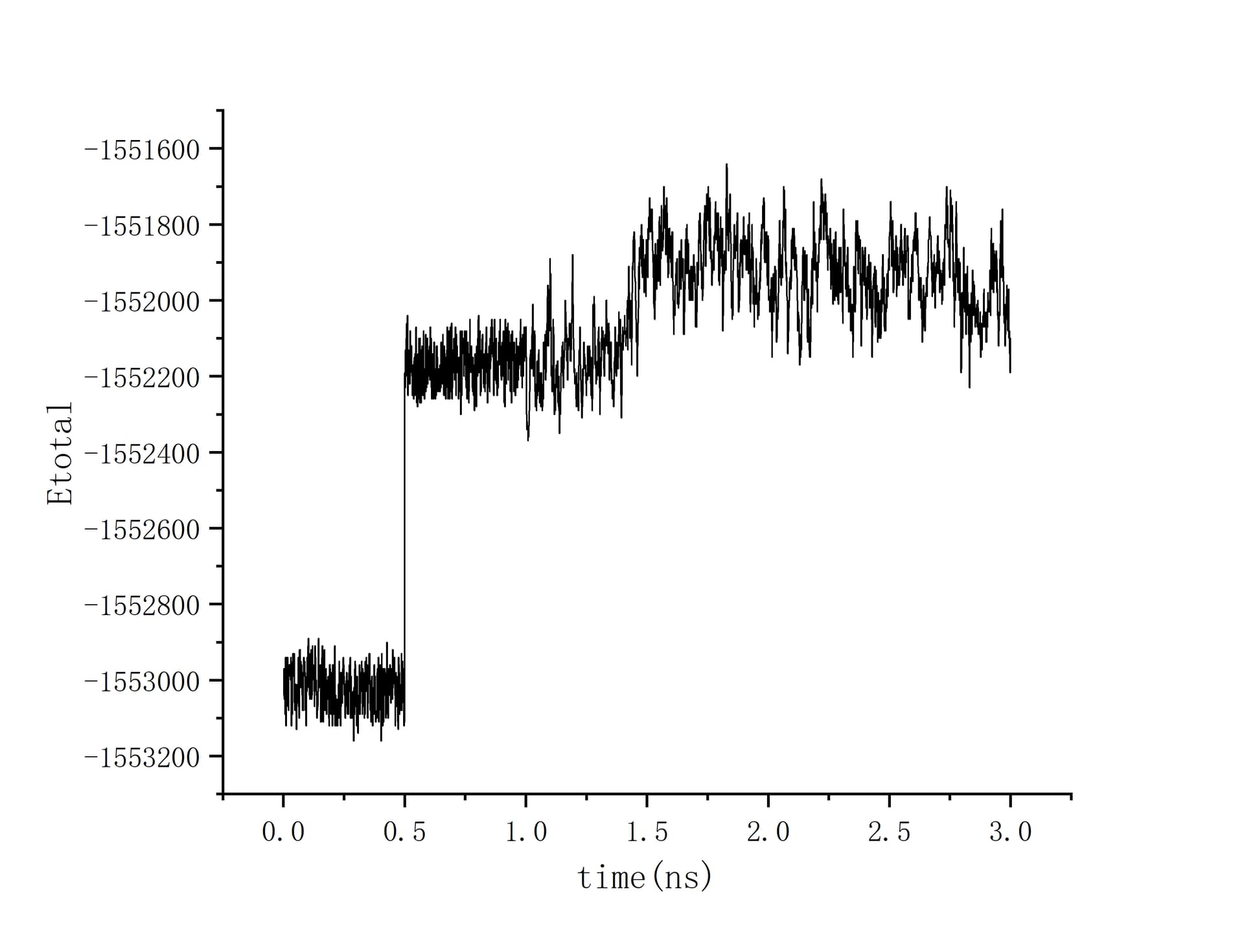
The total energy graph is the most self-explanatory piece of additional information. After the initial equilibration for the first 0.5ns, the total energy of the system jumps to a new average value from 0.5 to 1.0 ns, and then has the same average but larger fluctuations from 1.0 to 1.5ns, after which there is another jump in energy and still larger, slower energy fluctuations.
I am not sure how those correspond to either your changes in simulation parameters, or to some change in the condition of the PEO at particular times or temperatures. Beyond that, I don’t have more specific information or insight. For that, you should refer to the scientific literature for papers about similar studies.
I appreciate your prompt reply again.
The timestep is set as 1fs, and equilibration temperature is set as 305K. As the total energy graph illustrated, for the first 0.5ns, I relax the “all group” using the nvt ensemble at 305K, and for the second 0.5ns, I do the exactly same for the “main group”, where the main group is the part of the all group after the fixed layer is removed. This main explain the first jump at 0.5ns. And Starting at 1.0ns, I started taking the langevin hot bath as shown in the pasted script. The second jump at 1.0ns maybe is a sign that system is re-equilibrating. The total simulation script is pasted as below: