Hello. I am using LAMMPS (22 Jul 2025) on a Windows platform.
In a deformed simulation box, I want to move an atom along the x direction by a fixed distance.
Below is a simplified version of my input script:
variable rate equal 0.1
units lj
dimension 2
boundary p p p
atom_style atomic
lattice sq2 0.8
region box prism 0 71 0 71 -0.5 0.5 0.0 0.0 0.0
create_box 1 box
change_box all xy final $(0.48*ly) units box
create_atoms 1 single 73 106 0 units box
create_atoms 1 single 56 56 0 units box
group pinned_atoms id 1
mass 1 1.0
set group pinned_atoms image 0 -1 0 # or 0 0 0
timestep 0.005
write_dump all custom initial.dump id type x y z
fix ref all store/state 0 x y z
variable start_time equal $(step*dt)
variable strain equal "v_rate*(step*dt-v_start_time)"
variable x_aff atom "v_strain*f_ref[2]"
variable aff_vec atom "v_rate*f_ref[2]"
fix move_pinned pinned_atoms move variable v_x_aff NULL NULL v_aff_vec NULL NULL units box
run 1000
write_dump all custom tst_move.dump id type x y z f_move_pinned[1] f_move_pinned[2]
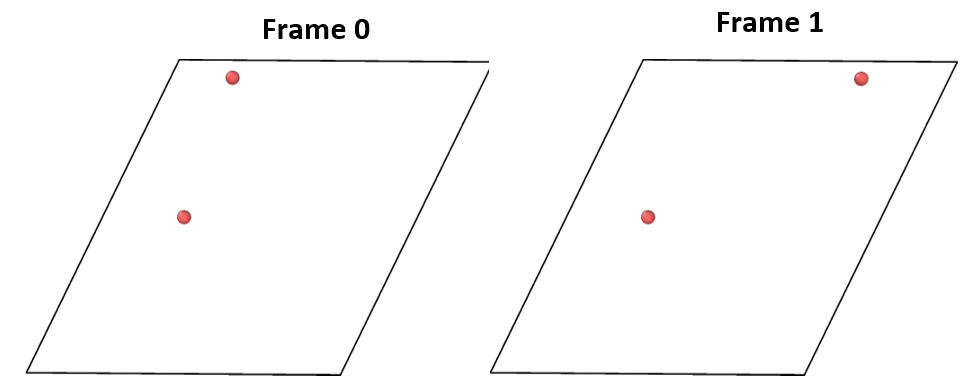
I found that when the image flag of the atom to be moved is set to 0 0 0, the behavior is as expected:
The top atom moves to a larger x position.
However, when I set the image flag of the atom to 0 -1 0, the behavior is not as expected:
The atom does not move forward, but instead moves backward.
I understand that fix move uses unwrapped coordinates, so when the image flag is 0 -1 0, the starting position is different.
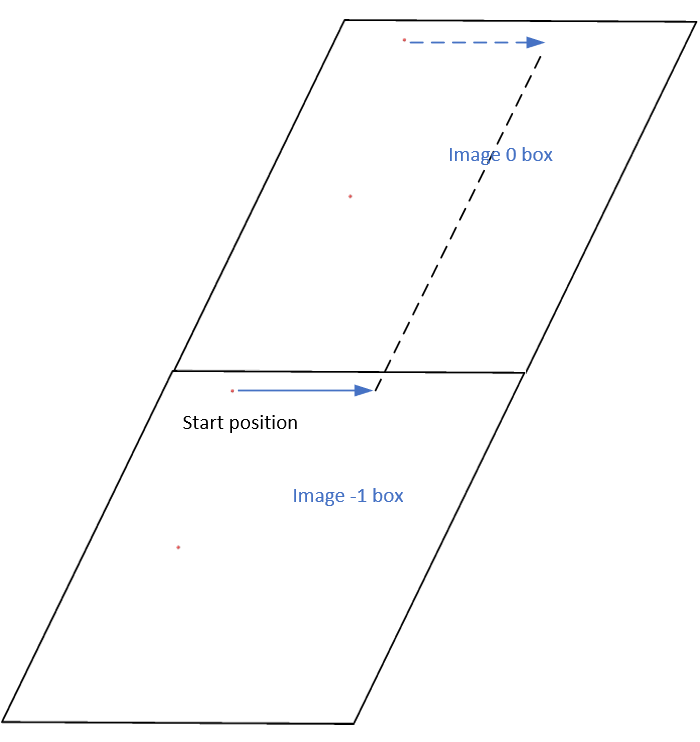
The starting position is located in the image -1 box rather than the image 0 box.
I suspect that your expectations are incorrect. Since you are using variables and not the “linear” mode, the question is really, whether you compute and feed to fix move what is commensurate with your expectations. From a cursory look it seems to me like your variable for the position update contains the position and not the displacement as described in the fix move documentation and that may be the origin of your confusion. Please also note, that your fix store/state stores the wrapped positions and not the unwrapped ones.
In my code, the displacements of particles are determined by the y - component of their vectors in the starting configuration. Specifically, atoms with a larger y - value experience a greater displacement.
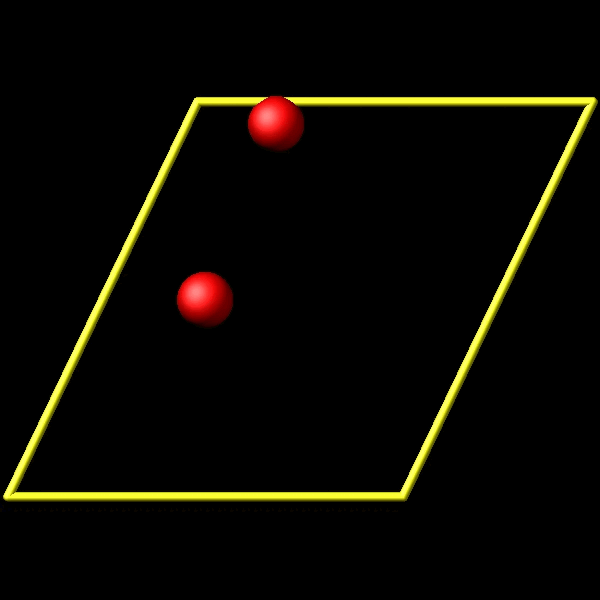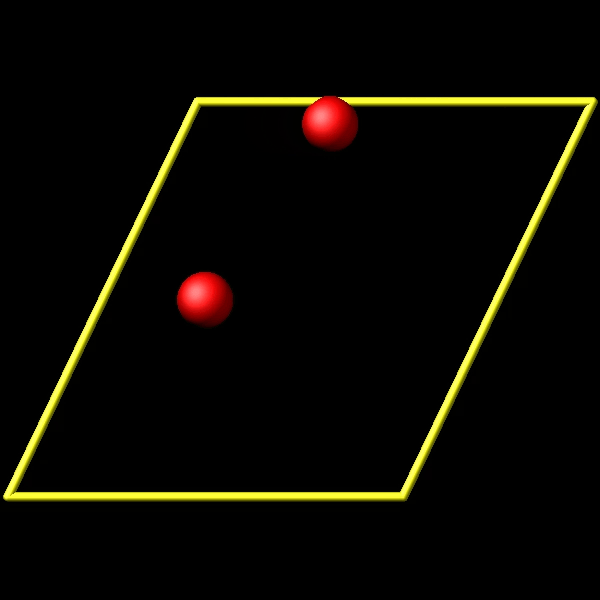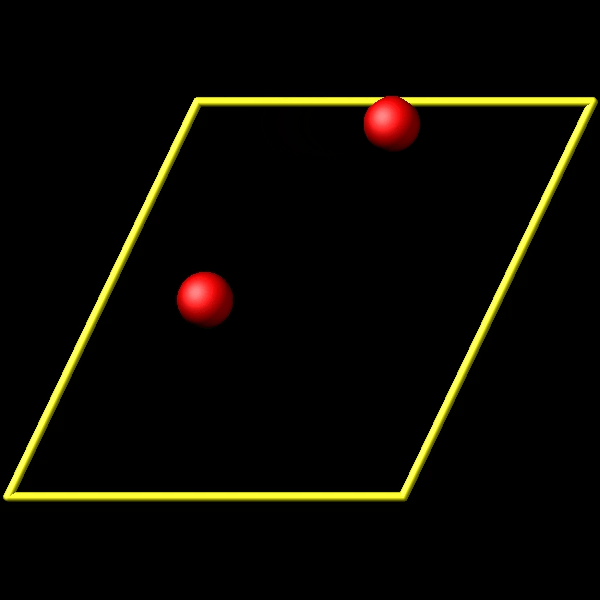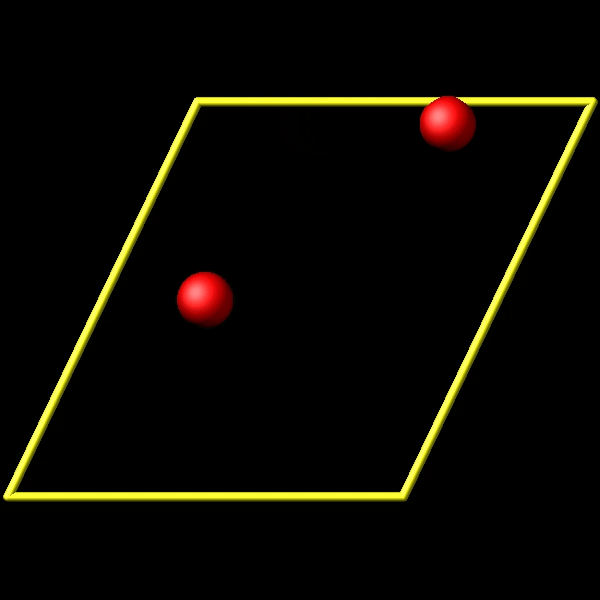
To address this, I utilize the fix store/state command to preserve the initial positions of the particles. However, this approach is incompatible with the “linear” style of the fix move command, which relies on an “equal - style” variable. Under these circumstances, the expression for the atomic displacements should incorporate the initial position information. As shown in the figures above, the fix move command moves atoms by a fixed distance regardless of the box in which the atoms are located.
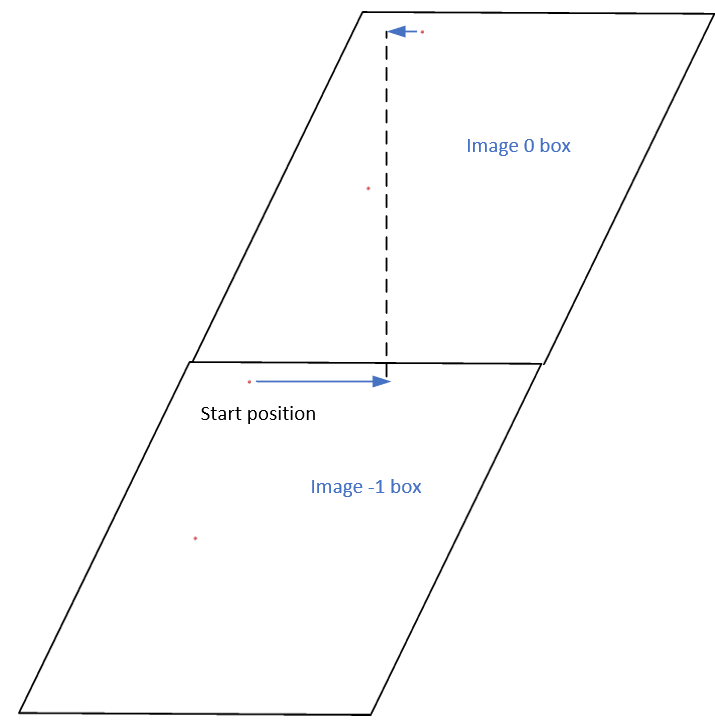
Since the fix move command stores unwrapped coordinates, the starting position should be in the image - 1 box. When an atom is remapped back to the origin (image 0) box, the mapping process should be consistent with the deformed box, which adheres to the periodic boundary condition (PBC) rule, in my opinion
No, it does not. Only the “variable” style uses variables. You likely are misinterpreting the following statement in the documentation:
Note that the linear style is identical to using the variable style with an equal-style variable that uses the vdisplace() function
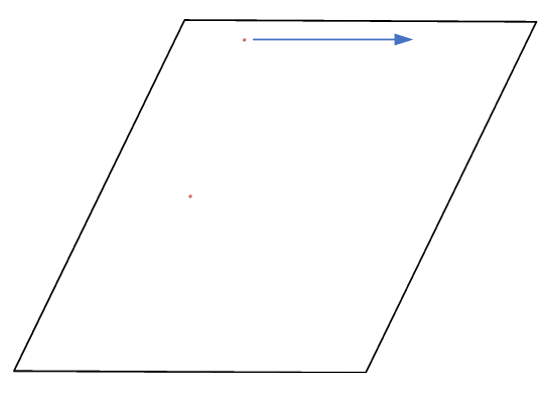
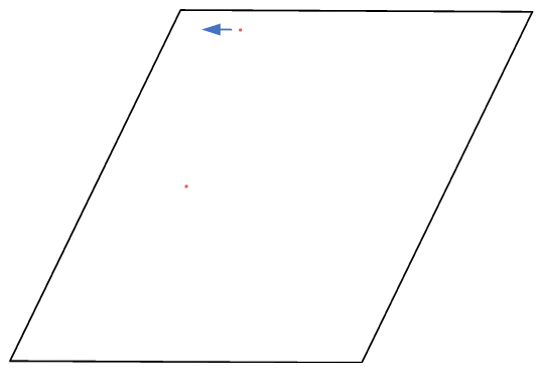
Actually, it does move forward. Only when you look at it, it has passed through the box and wrapped around periodic boundaries. See below for animations of your fix move applications. On the left for the 0 0 0 image flag case, and on the right for the 0 -1 0 image flags case.
Thank you very much. I sincerely appreciate your input. I’ll carefully examine the documentation regarding the fix move command.
When I refer to the “first” or “frame 0”, I’m specifically talking about the time order that can be obtained from the dump command.
Below, I’ve attached my updated input file where I’ve added the dump command.
variable rate equal 0.1
units lj
dimension 2
boundary p p p
atom_style atomic
lattice sq2 0.8
region box prism 0 71 0 71 -0.5 0.5 0.0 0.0 0.0
create_box 1 box
change_box all xy final $(0.48*ly) units box
create_atoms 1 single 73 106 0 units box
create_atoms 1 single 56 56 0 units box
group pinned_atoms id 1
mass 1 1.0
set group pinned_atoms image 0 -1 0 # or 0 0 0
timestep 0.005
write_dump all custom initial.dump id type x y z
fix ref all store/state 0 x y z
variable start_time equal $(step*dt)
variable strain equal "v_rate*(step*dt-v_start_time)"
variable x_aff atom "v_strain*f_ref[2]"
variable aff_vec atom "v_rate*f_ref[2]"
fix move_pinned pinned_atoms move variable v_x_aff NULL NULL v_aff_vec NULL NULL units box
dump record_motion all custom 10 move_process.dump id type x y z f_move_pinned[1] f_move_pinned[2]
run 1000
write_dump all custom tst_move.dump id type x y z f_move_pinned[1] f_move_pinned[2]
In my particular situation, frame 0 corresponds to Timestep 0, and frame 1 corresponds to Timestep 10.
I don’t have the time to debug your input for you. With using the variable option you get the positions that you feed fix move… If those are not what you want, the fault is not with LAMMPS.
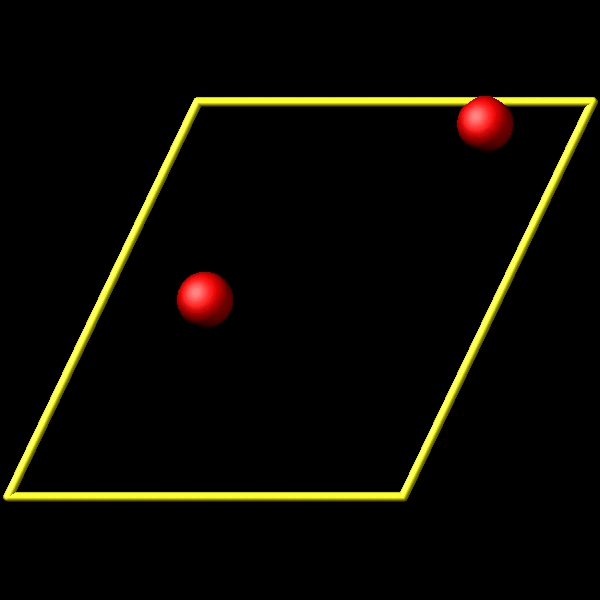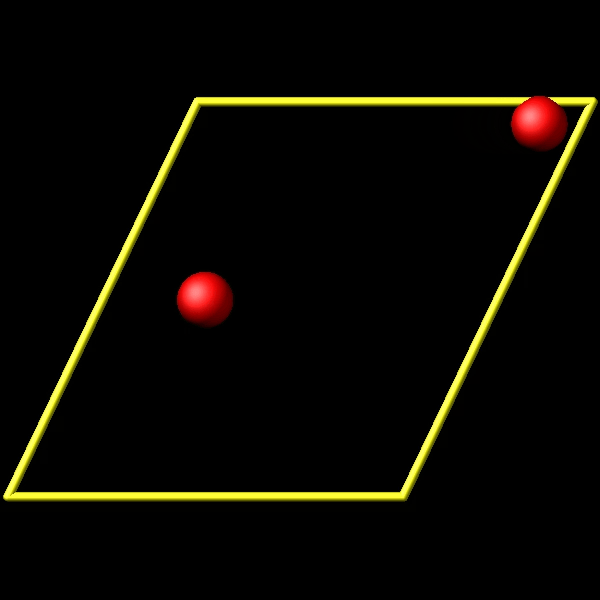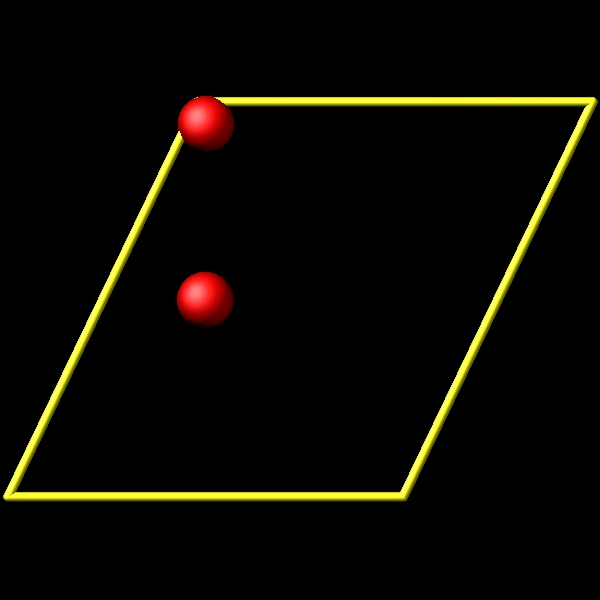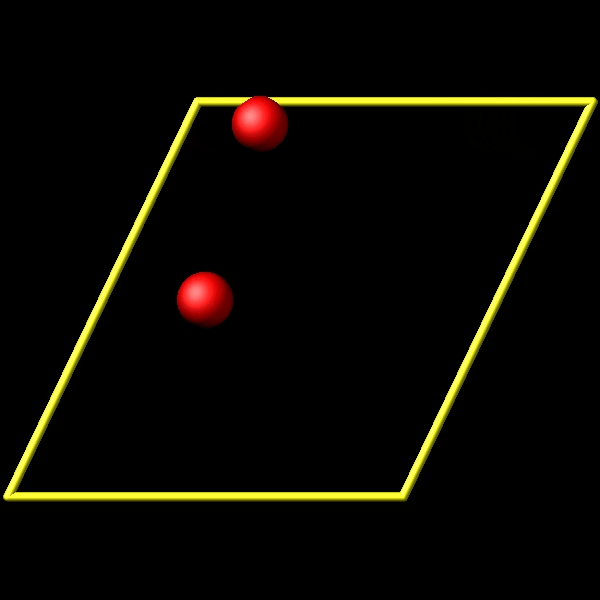 .
.