Hello, I have this error while running. After I searched on the Internet, some people said that increasing the number of iterations and adjusting the tolerance can be solved, but I did and did not solve, I would like to ask what should be done now.


First, you should post to the correct category. materials project is obviously not about LAMMPS. I have found your post by accident when looking for something else and recategorized it. Don’t expect this to happen always.
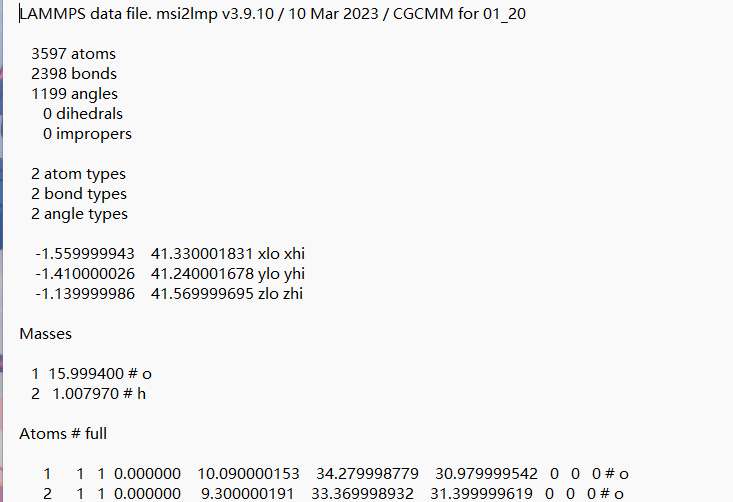
Second, your data file has bonds and angles, but that is wrong for ReaxFF. In ReaxFF those are all implicit.The bonded interactions in the data file will lead to exclusions in the non bonded part and that will mess up your force field in a bad way. Basically what you are computing is totally bogus.
First,this is a problem that I’m having with lammps. Can you tell me why this problem is not about lammps?
Second, if I don’t set angle type and bond type then lammps will report an error when I run it. The error is that I don’t set either angle type or bondtype.
I just want to know what’s causing“Fix qeq/reaxff/OMPP…”
Exactly, but you didn’t post to a
LAMMPS category. Instead you posted your inquiry in the “Materials Project” category, which is not about LAMMPS. I recategorized it for you to the LAMMPS category of this server. MatSci hosts forums for many different projects and software packages.
You are not paying attention. Your main problem is with the data file. It should not contain any bonds or angles sections or bond or angle types. Once those are removed, you do not need to specify a bond or angle style. And this is how ReaxFF calculations work. There are not explicit bonds or angles, they are all implicitly defined (and dynamically redefined during a run) by the reaxff pair style.
The QEq failure is an indirect consequence of the presence of bonds and angles, as it will cause exclusions in the pair style neighbor lists, which will make the calculation unphysical.