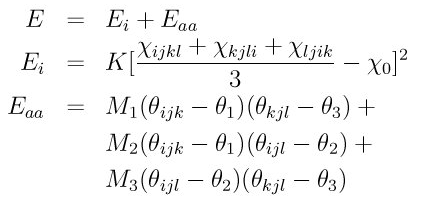Dear lammps users,
I am trying to transfer a force field described in GULP to lammps, and i run MD simulation with the same parameters in both the two package, using the same post pocessing tool, but the result spectrum is very different, see the following two spectrum, the one on the right is from GULP, and the other one from lammps. i really don’t know what will be the reason for the difference. someone please help me. thanks very much!!!
Best Regards
Jiasen Guo
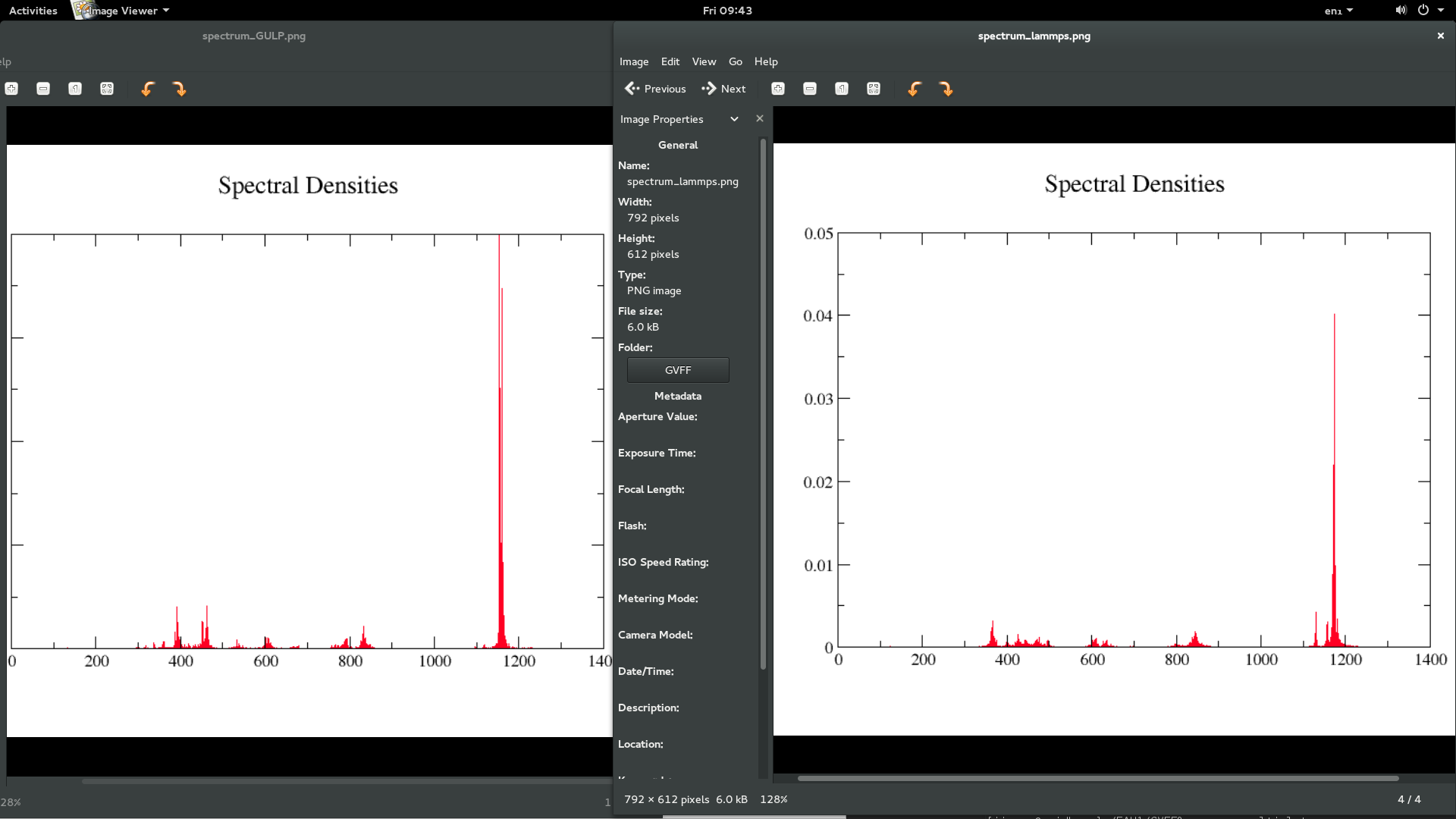
the force field specification in GULP follows,
harmonic bond
Si O 33.57535587 1.61
three bond
Si O O 4.8678025239 109.47
O Si Si 0.748892696 149.8
bcross bond
Si O O 1.1857467686 1.61 1.61 0 1.8 0 1.8 0 3.6
O Si Si 1.5601931166 1.61 1.61 0 1.8 0 1.8 0 3.6
bacross bond
Si O O 0.8113004207 0.0000 1.61 1.61 109.47
xangleangle bond
Si O O O -0.6864849713 -0.6864849713 -0.6864849713 109.47 109.47 109.47
I attach the GULP doc for specific terms, hoping it helps you understand the way GULP defines force field.
bond cross term

bond angle cross term

angle cross term

the force field specification in lammps follows,
bond_style harmonic
bond_coeff 1 16.787678 1.61
angle_style class2
angle_coeff 1 109.47000 2.4339013 0.00000 0.0000
angle_coeff 1 bb 1.1857467686 1.61 1.61
angle_coeff 1 ba 0.8113004207 0.0000 1.61 1.61
angle_coeff 2 149.80000 0.3744463 0.00000 0.0000
angle_coeff 2 bb 1.5601931166 1.61 1.61
angle_coeff 2 ba 0.000000000 0.0000 1.61 1.61
improper_style class2
improper_coeff 1 0.0 100
improper_coeff 1 aa -0.6864849713 -0.6864849713 -0.6864849713 109.47 109.47 109.47
pair_style coul/long 12.0
pair_coeff * *
kspace_style ewald 1.0e-4
special_bonds coul 1.0 1.0 1.0
details from lammps doc.
angle_style class2
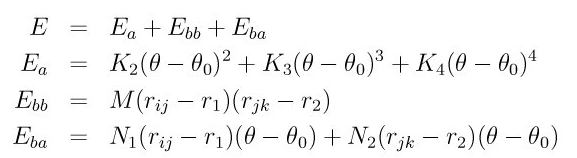
improper_style class2