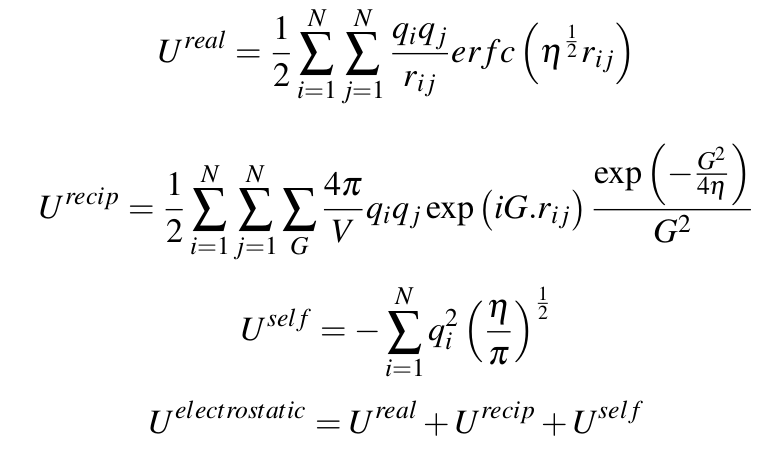Hi, all
thanks for the reply of steve. i did a simple test with a same molecular configuration, with only electrostatic potential. see the following output from GULP and lammps. the energy in this case differs by 0.0005 ev, (actually in the previous email, i report the energy difference between GULP and Lammps calculated on another molecular configuration is 0.05 ev, this is a typo, i am sorry for that, it should be 0.0005 ev).
LAMMPS
Memory usage per processor = 16.9445 Mbytes
---------------- Step 0 ----- CPU = 0.0000 (sec) ----------------
TotEng = -58.447991 KinEng = 0 Temp = 0
PotEng = -58.447991 E_bond = 0 E_angle = 0
E_dihed = 0 E_impro = 0 E_vdwl = 0
E_coul = -29.219132 E_long = -29.228859 Press = -43321.939
Volume = 720.52803
GULP
Components of energy :