Hi, everyone,
i have a question about how to build my integrator using python.
my team’ve develop a new integration algorithm and want to make it by lammps python api. therefore, to build a framework for algorithm migration, we firstly use python package of lammps to implement velocity-verlet integrator (VV) (lammps integrator by default).
the VV integrator made by ourselves works under NVE ensemble but does not work under NVT ensemble. here are the core function.
def gradientFunction(Lammps: IPyLammps | PyLammps, Position: np.ndarray) -> np.ndarray:
"""
define the gradient function
"""
llnp = Lammps.lmp.numpy
x: np.ndarray = llnp.extract_atom('x')
x[:] = Position
before_id = llnp.extract_atom('id').copy()
Lammps.run(0, 'pre yes post no');
after_id = llnp.extract_atom('id')
assert (before_id == after_id).all(), 'array has changed !!!!!'
force: np.ndarray = llnp.extract_atom('f')
return force.copy()
def VelocityVerlet(Lammps: PyLammps | IPyLammps, basis_timestep: float, ntimestep: int, disable_tqdm: bool = False):
def excute(Lammps: PyLammps | IPyLammps, Dt: float):
# get the initial state of system
lmpX, lmpV = Lammps.lmp.numpy.extract_atom('x'), Lammps.lmp.numpy.extract_atom('v')
lmpMass, atomtype = Lammps.lmp.numpy.extract_atom('mass'), Lammps.lmp.numpy.extract_atom('type')
# copy variables
x_0, v_0 = Lammps.lmp.numpy.extract_atom('x').copy(), Lammps.lmp.numpy.extract_atom('v').copy()
mass = rearrange(lmpMass[atomtype], "l -> l 1")
# keep the atoms of boundary stationary
matrix = np.ones_like(x_0)
matrix[atomtype < 3] = 0.
v_0[atomtype < 3] = 0.
ftm2v = ftm2v_coeff[Lammps.system.units]
massinv = matrix * ftm2v / mass
Dt_half = Dt * 0.5
# get gradient at t by using Lammps API and compute v(t + Dt/2)
force = gradientFunction(Lammps, x_0)
v_ht = v_0 + massinv * force * Dt_half
# compute r(t + Dt)
x_t = x_0 + v_ht * Dt
# get gradient at t + Dt by using Lammps API and compute v(t + Dt)
force = gradientFunction(Lammps, x_t)
v_t = v_ht + massinv * force * Dt_half
# put x_t, v_t into source address
lmpX[:] = x_t; lmpV[:] = v_t
with tqdm(total = ntimestep, desc = 'vv Iteration: ', leave = False, position = 1, disable = disable_tqdm) as vv_bar:
for _ in range(ntimestep):
excute(Lammps, basis_timestep)
vv_bar.update()
return
here are the lammps code
def create_simulation(timestep = 0.2, cmdargs = None, num_threads: int = 1, ensemble: str = 'nve') -> IPyLammps:
lmp = lammps(cmdargs=cmdargs)
MDsimulation = PyLammps(ptr = lmp)
MDsimulation.enable_cmd_history = True
if num_threads > 1:
MDsimulation.package(f"omp {num_threads} neigh yes")
MDsimulation.suffix('omp')
MDsimulation.units('real')
MDsimulation.atom_style('full')
MDsimulation.pair_style('lj/cut/coul/long 12.0')
MDsimulation.bond_style('harmonic')
MDsimulation.angle_style('harmonic')
MDsimulation.dihedral_style('opls')
MDsimulation.improper_style('cvff')
MDsimulation.dielectric(1.0)
MDsimulation.pair_modify('mix arithmetic')
MDsimulation.special_bonds('lj/coul 0.0 0.0 1.0')
MDsimulation.read_data('input/data.lammps')
MDsimulation.atom_modify('sort 0 0.0') # turn off sort algorithm
MDsimulation.set('type 1 charge -0.55')
MDsimulation.set('type 2 charge 1.1')
MDsimulation.group('zeo type 1 2 ')
MDsimulation.group('indole type 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18')
MDsimulation.kspace_style('pppm', 1e-4)
MDsimulation.neighbor('10.0 bin')
MDsimulation.neigh_modify('every 1 delay 0 check yes exclude molecule/intra zeo')
MDsimulation.delete_bonds('zeo multi')
MDsimulation.velocity('indole create 700.0 902144 dist gaussian')
MDsimulation.velocity('zeo set 0.0 0.0 0.0')
MDsimulation.velocity('indole scale 700.0')
if ensemble == 'nvt':
MDsimulation.fix('1 indole nvt temp 700.0 700.0 1.0')
# MDsimulation.fix("1 indole temp/rescale 1 700.0 700.0 0.01 1.0")
# MDsimulation.fix("2 indole nve")
elif ensemble == 'nve':
MDsimulation.fix('1 indole nve')
MDsimulation.compute('1 indole temp')
MDsimulation.thermo_modify('lost/bond ignore')
MDsimulation.thermo_style("custom step time spcpu temp press pe ke enthalpy evdwl emol ecoul etotal")
MDsimulation.timestep(timestep) # attn set timestep
# initialize system state
MDsimulation.run(0, 'pre yes post no')
return MDsimulation
here 's the energy variation graph over time in the NVE ensemble. it works.

and here’s energy variation graph over time in the NVT ensemble.
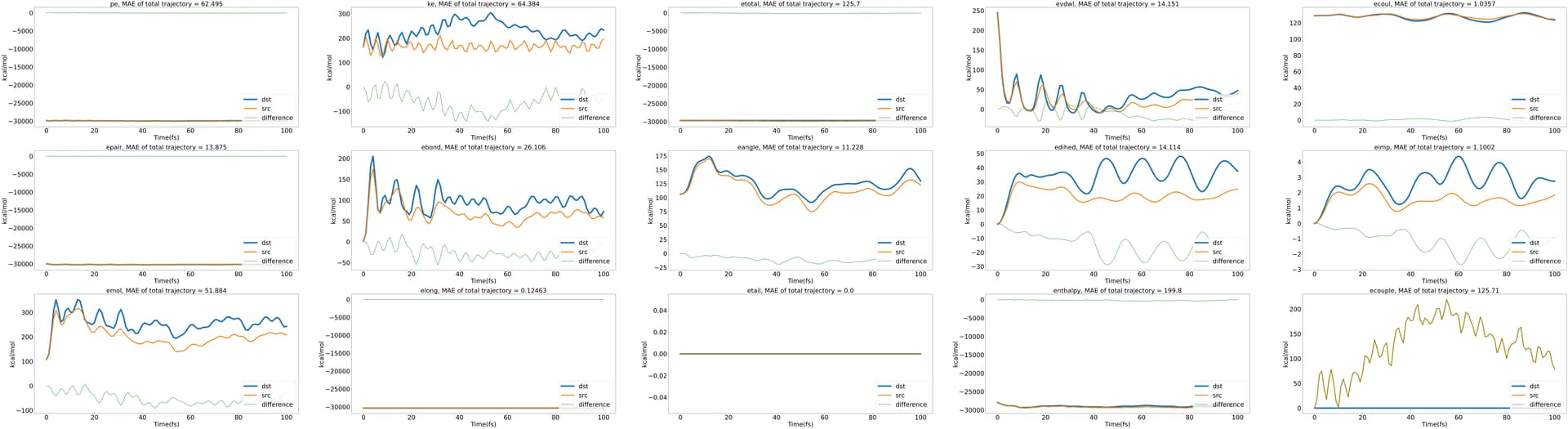
how can i implement the function for correct data?
Thank you so much