Dear Lammps users/developers,
I want to simulate a particle containing formic acid and water, and the NPT ensemble is used. The formic acid molecule is fully rigid. I use the method recommended in the manual. Therefore, two kinds of “in” files are listed:
①
fix a2 formic rigid/npt molecule temp 180 180 100 iso 1.0 1.0 1000 dilate all
fix shk water shake 0.0001 50 100 b 5 a 5 t 6 7
fix b2 water nvt temp 180 180 100.0
②
fix shk water shake 0.0001 50 100 b 5 a 5 t 6 7
fix b2 water npt temp 180 180 100.0 iso 1.0 1.0 1000 dilate all
fix a2 formic rigid/nvt molecule temp 180 180 100
I get two different results, of which the first crashes after hundreds of thousands of steps, and the second method can operate normally. The change of system’s pressure have been tracked and stated as follow.
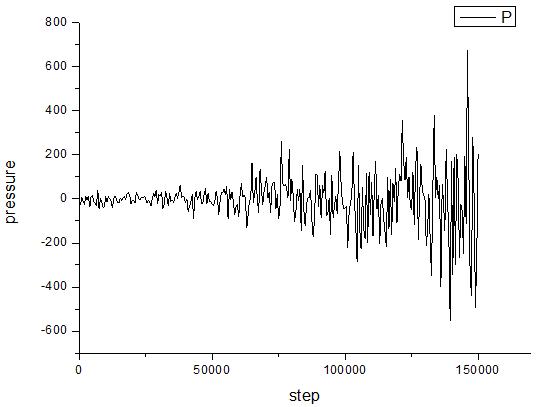
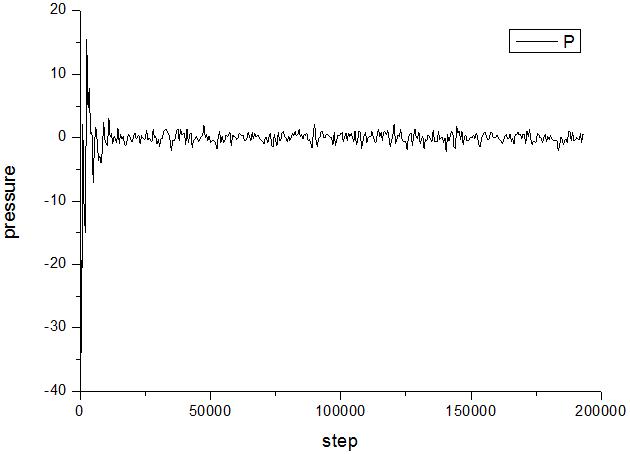
a. Method ① b. Method ②
When I tack the volume of the system, I have found that the volume of method ① decreases monotonically from a=8nm to a=3.8nm. Then the simulation crashes. In addition, when the Pdamp is changed from 1000 to 2000, the result remains the same. However, the volume of method ② increases from a=8nm to about a=22nm, and the error is not occurring.
I am confused by two question: ⑴ why results of these two methods is different, I have learnt from the manual that a global pressure or stress tensor is computed for all atoms, regardless of what atoms are in the fix group.⑵ why the simulation crashes when method ① is used. I want to know whether the reason is the wrong use of fix npt command in the “in” file or the wrong physical model.
I would appreciate any help and suggestion.
Thanks!
Zhang Chao