Dear Everyone,
I am wondering if Lammps can capture polymer entanglements effect?
I want to increase the length of my polymer chain so that there will be entanglement between chains. Then I will do uni-axial loading on the polymer system. I am not sure what command I should use.
Thank you very much for any comments,
Lili Zhang
sure - search the LAMMPS papers page in
your browser and the word "entanglement"
appears in several papers. Also see the Pictures
page for work by Mark Robbins group on pulling
apart a polymer mixture. The fix deform command
can do that.
Steve
If you are wondering how to choose the initial coordinates of the
polymers, this is not trivial. Definitely LAMMPS does not a have a
built-in command to generate these kinds of structures. Pizza.py has
a chain command to build melts:
http://pizza.sandia.gov/doc/chain.html
and ESPresSo has a similar "polymer" command which does something
similar (but that does not work with LAMMPS).
I would definitely take a look at the papers Steve mentioned, to see
how they do it.
One of the more rigorous methods I know of was written by Marc L.
Mansfield. In a number of (somewhat older) papers describing a
monte-carlo algorithm to generate a fully-equilibrated random melt of
polymers. (Last I checked, these algorithms generate "lattice"
polymers: each bead occupies a site on a cubic lattice.) The initial
conformations generated by these algorithms are guaranteed to be
compact, random and entangled. I don't know how complicated your
polymers are. I don't know which of his papers are the best ones.
Anyway here are a few links:
http://jcp.aip.org/resource/1/jcpsa6/v77/i3/p1554_s1
http://jcp.aip.org/resource/1/jcpsa6/v133/i4/p044903_s1
To my knowledge, I don't think any of the molecule building programs
use these kinds of algorithms, but the algorithm is not hard to
implement yourself. Probably the "chain" or "polymer" tools above
would work well enough.
I have no idea if this is relevant to what you were asking.
Cheers!
Andrew
Or you could ask Joerg Rottler (http://www.phas.ubc.ca/~jrottler/) how
he did it. He's worked extensively with Mark Robbins on the topic and
is also a LAMMPS user. I'm sure he'll be happy to comment provided you
put together a precise and objective message.
Carlos
I believe packmol will build entangled polymers if you create “ring-shaped” oligomers to feed to packmol.
I created relatively flat oligomers that were like a “square” ring where I used four curing molecules
as the corners of the square and five polymer molecules to connect them - one for each side with one side getting two.
Packmol packed them into a box. I expected the oligomers to be laid flat on one another but they were “entangled” such that the
tolerance between atoms of different molecules was honored. My packmol created MD cell looked
amorphous to me. Perhaps one could create a polymer chain that is very twisty and by using packmol one could populate an
MD cell with many of these polymers to get the necessary entanglement? I annealed my packmol MD cell into an MD cell with
the desired density. At some point in time a piece of vaporware I am working on will do entanglement too.
That would be awesome.
Andrew
Dear All,
Thanks for your comments.
I have been using "fix deform " command to do uni-axial tension on the polymers where the length of the chain is less than the entanglement length. No problem there.
Now I have Packmol and Moltemplate working which enable me to construct longer chains to have the chains entangled.
The material response is expected to be different due to these entanglements during the deformations, since one chain can’t pass through the other chains at the entanglement points. Please see the figure attached.
In the “fix deform” doc page I didn’t find any thing to clarify that this command take the entanglement into account. So that during simulation lammps checks the chains entanglement regions to stop one polymer chain pass through the others at the this region.
Thanks,
Lili Zhang
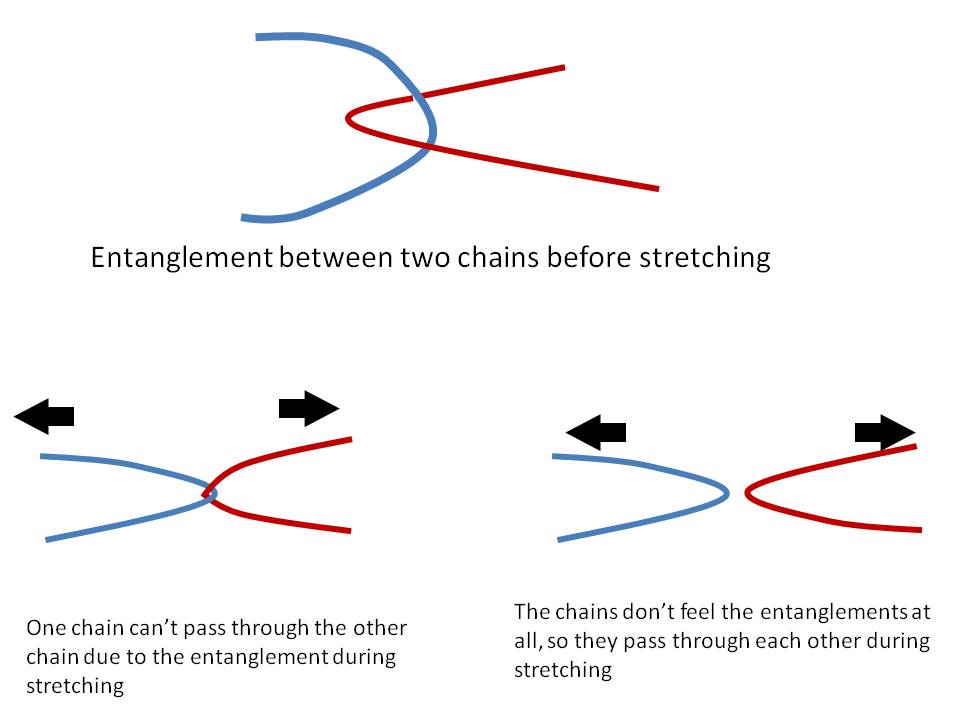
Can you clarify what you mean? ("take the entaglement into account")
(The repulsion between atoms should prevent chains from passing
through each each other.)
Cheers
Andrew
How long does your polymer need to be to see entanglement? How many polymers do you want to put into a box?
I have been building models of oil shales as 3d cross linked hydrocarbons using a proximity embedding algorithm to build a 3d structure starting from a linear polymer. Crosslinking is done in a second step based on the proximity of chain ends to aromatic centers. I have also used this algorithm to build boxes of solvents, and oligomers. For example can I build a box of 50 chains of a 30 mer of polyethylene oxide where the chains are entangled, the orientations are random and the internal dihedral angles are random. The code has been published as part of a smiles to 3d converter I wrote several years ago (smi23d) and I think it is still available on sourceforge. It basically takes a connectivity table and turns it into a set of 3d coordinates. In the smi23d code the connectivity table was built from the smiles string but you can replace this with any structure reading code that can generate a connectivity list (I used Sybyl mol2 files in my current work). I am attaching the peo_30 cluster file fyi.
Kevin
peo30_cluster_opt.mol2 (398 KB)
Lili,
If you are using atomistic or hard-core potentials (like K-G), then there is no need to explicitly prevent chain crossings… as Andrew said, the energy barrier is very high. Chain crossing is a problem tied to soft coarse-grain potentials used with a big timestep.
cheers
Tim
Hi, Lili. Do you want to do entanglement analyses? It is easy to do various types of primitive path analysis in LAMMPS. See e.g. Phys. Rev. E, v. 72, article 061802 (2005) for a description of the necessary equilibration methods and Phys. Rev. E, v. 80, article 031803 (2009) for a description of how long your chains need to be to be well entangled. The key to doing PPA is to disable intrachain excluded volume interactions while retaining interchain EV. This can be done using the neigh_modify exclude molecule all command.
Best,
Rob
Dear Rob and Everyone,
Yes, I want to see how the polymer properties (e.g. stress- strain response) change with different molecular length. The entanglement will inevitably appear when the chain length gets longer. The force field I use is PCFF.
Just want to be clear, the only thing I need to add to the non-entanglement case is the " neigh_modify exclude molecule all " command?
Thanks for the papers recommended, I will check them out.
Lili
Hi. No, neigh_modify exclude molecule all is just for primitive path analysis (see Everaers et al, Science, v. 303, p 823 (2004)) which measures the entanglement length N_e. It disables intrachain excluded volume, so you wouldn’t want to use it for a normal run! It wouldn’t get rid of the entanglements, only reduce your chain contours to primitive paths.
Best,
Rob