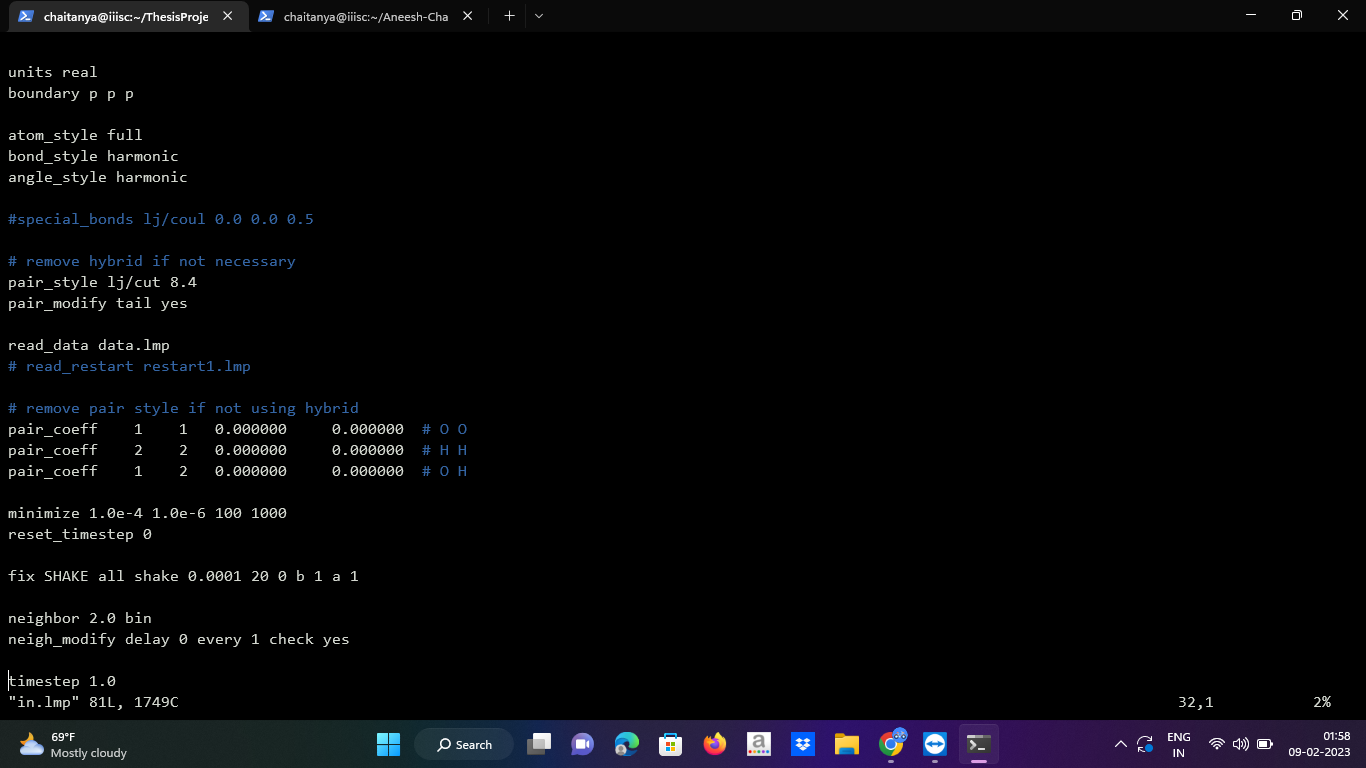
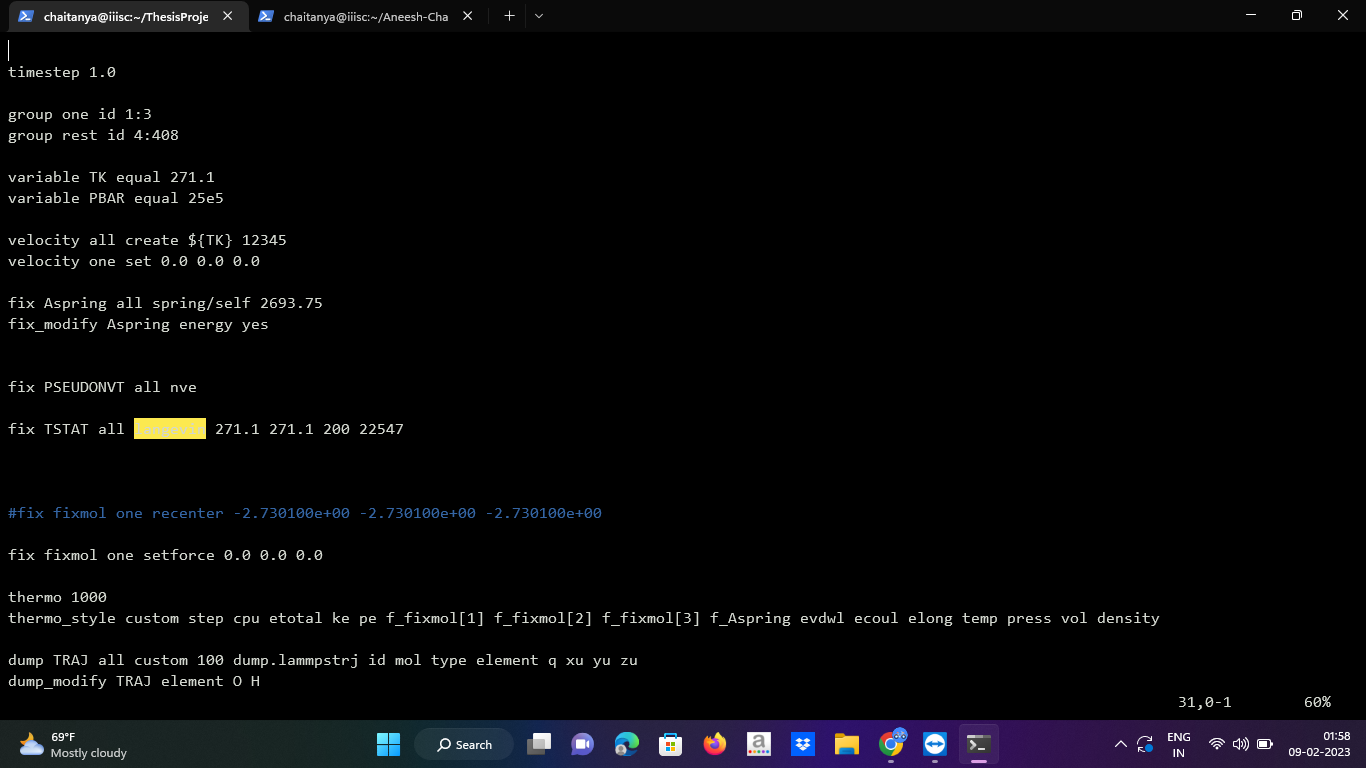
I am trying to do free energy calculations of ice for which I am following the Einstein molecule method. For this I need to attach each atom of all the water molecules to springs, which I am doing using spring/self command. Furthermore, I need to fix the oxygen atom of one of the molecules in my system. To do so, I am using the ‘setforce’ fix and setting the initial velocity to zero for the atom chosen to be fixed (i.e. freeze).
But, I get an error saying atoms are lost. I am unable to understand what is going wrong in my system. Although I should mention that each molecule does not interact with each other, i.e. these are ideal gas like molecules, which experience only spring force.
The timestep I am using is 1 fs. The error reads:
ERROR: Lost atoms: original 408 current 3 (…/thermo.cpp:439)
{I don’t understand if the current atom is the number of atoms lost or remaining}
The simulation runs without any error if I reduce the timestep, but I do not want to do that as I have to run a really long simulation. Also, I do not understand why should atoms be lost if there is a spring attached with a substantial spring constant of 2700 kcal/A^2.
Please explain why it happens and what is the solution apart from reducing the timestep.
I am attaching my input file screenshots.
Uploading screenshots is a waste of bandwidth and is making it a much bigger effort to check out your input compared to attaching the file in.melt (736 Bytes)
or inserting the text as quoted text (using triple backquotes (```)) as shown below for the “melt example”. One cannot easily cut-n-paste text from images.
If you want the best help, you need to make it easy to help you. The more effort it is to read and test what you are doing, the less likely is it that people will respond (and that is an exponential or worse relation).
# 3d Lennard-Jones melt
units lj
atom_style atomic
lattice fcc 0.8442
region box block 0 10 0 10 0 10
create_box 1 box
create_atoms 1 box
mass 1 1.0
velocity all create 3.0 87287 loop geom
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0 2.5
neighbor 0.3 bin
neigh_modify every 20 delay 0 check no
fix 1 all nve
#dump id all atom 50 dump.melt
#dump 2 all image 25 image.*.jpg type type &
# axes yes 0.8 0.02 view 60 -30
#dump_modify 2 pad 3
#dump 3 all movie 25 movie.mpg type type &
# axes yes 0.8 0.02 view 60 -30
#dump_modify 3 pad 3
thermo 50
run 250
A high spring constant means atoms vibrate with a high fundamental frequency (\omega \sim \sqrt{k/m}), which means you need a shorter integration timestep to accurately integrate their motion. Typically ten timesteps per oscillation is a good starting point (you still need to double check).
A quick order-of-magnitude estimate suggests that your fundamental oscillation period would be on the order of 1fs, which suggests that your timestep would need to be on the order of 0.1fs.
Sorry for the inconvenience. I am attaching the unit file as text.
boundary p p p
atom_style full
bond_style harmonic
angle_style harmonic
pair_style lj/cut 8.4
pair_modify tail yes
read_data data.lmp
pair_coeff 1 1 0.000000 0.000000 # O O
pair_coeff 2 2 0.000000 0.000000 # H H
pair_coeff 1 2 0.000000 0.000000 # O H
minimize 1.0e-4 1.0e-6 100 1000
reset_timestep 0
fix SHAKE all shake 0.0001 20 0 b 1 a 1
neighbor 2.0 bin
neigh_modify delay 0 every 1 check yes
timestep 0.8
group one id 1:3
group rest id 4:408
variable TK equal 271.1
variable PBAR equal 25e5
velocity all create ${TK} 12345
velocity one set 0.0 0.0 0.0
fix Aspring all spring/self 2693.75
fix_modify Aspring energy yes
fix PSEUDONVT all nve
fix TSTAT all langevin 271.1 271.1 200 22547
fix fixmol one setforce 0.0 0.0 0.0
thermo 1000
thermo_style custom step cpu etotal ke pe f_fixmol[1] f_fixmol[2] f_fixmol[3] f_Aspring evdwl ecoul elong temp press vol density
dump TRAJ all custom 100 dump.lammpstrj id mol type element q xu yu zu
dump_modify TRAJ element O H
restart 100000 restart*.delta_A1
run 1250000
write_data data_delta_A1.eq.lmp
Or reduce the spring constant. I do not know your method (Einstein crystals), so I can’t advise you on what works and doesn’t for your method. Good luck!
You could speed up your computation substantially by using pair style zero and picking a much shorter cutoff. Possibly, even pair style none could work. In that case, you have to set the communication cutoff appropriately via comm_modify.
It just needs to be long enough that the neighbor list code finds all bond and angle atoms for ghost atoms from the neighboring subdomains.
Perhaps 3 angstroms may suffice. LAMMPS will print a warning if it is too short.