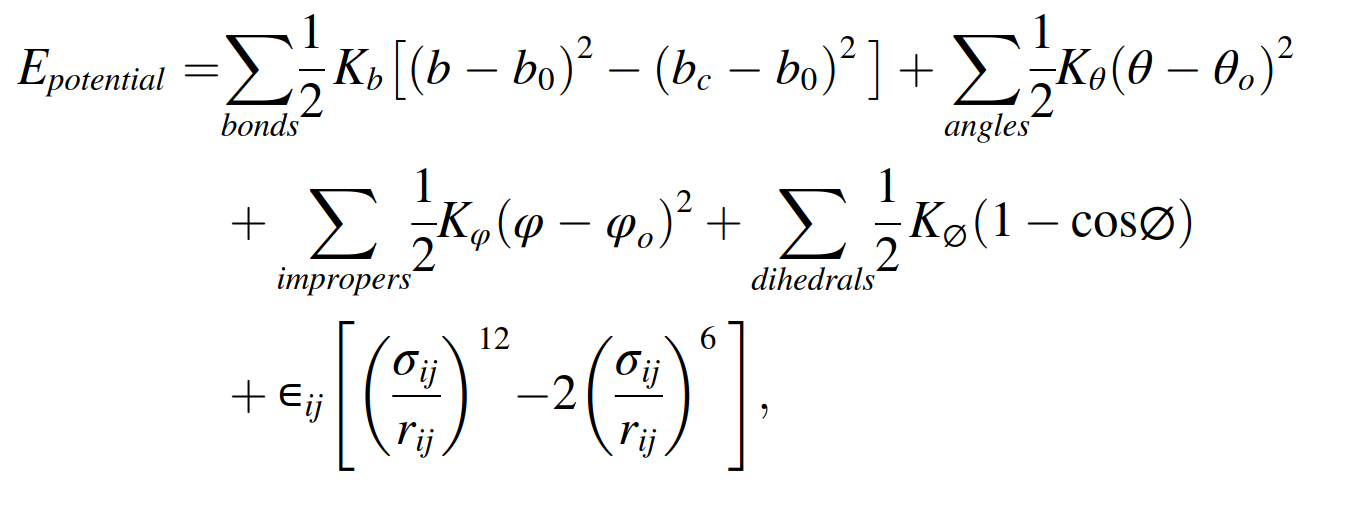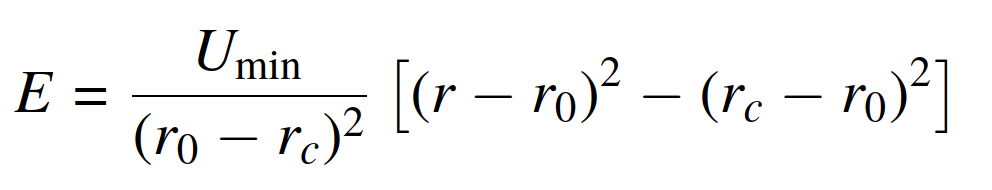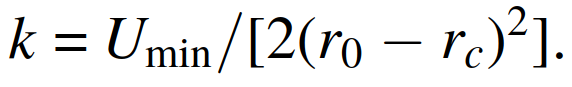, and I need to convert the force constant given in the paper to the corresponding coefficients used in the lammps code. From the lammps documentation(release 19Nov2024) section 5.11.3, it states that the harmonic/shift bond potential expression is
, then it states that the spring constant is
, which confuses me. According to my understanding, shouldn’t the spring constant be “k = 2Umin/(r0 − rc)^2” rather than “k = Umin/[2(r0 − rc)2]”? Is it a typo or something else?
Thank you very much for your time and your help are much appreciated.
Unfortunately, I don’t have something like a bond table to compare the bond_write output data with, because the paper only gives the force constant and final simulation results without any supplement materials or any other kind of related data.
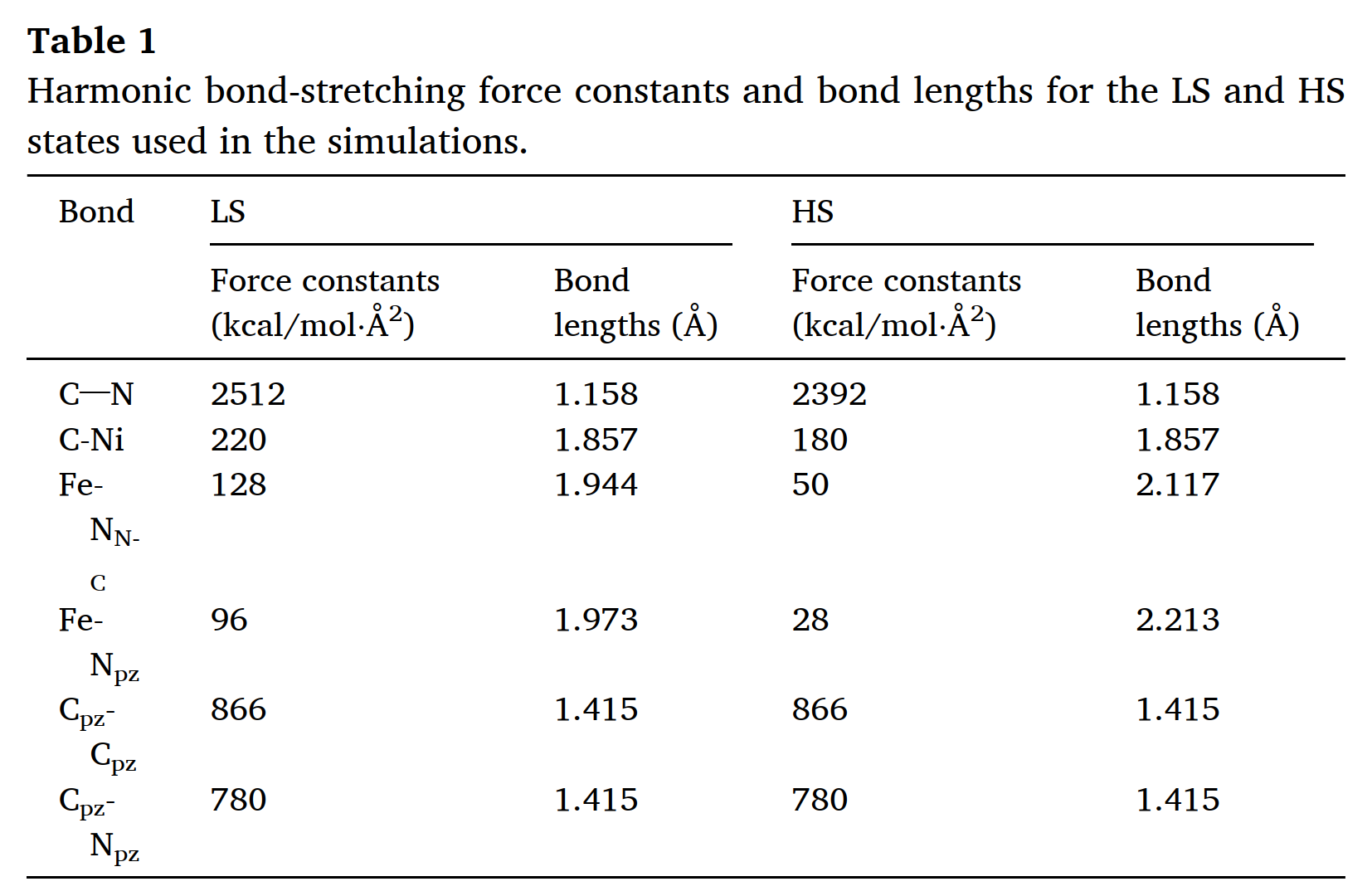
The reason I want to know if the k expression in the lammps documentation was a typo is that I’m on the way to figure out the cause of the discrepancy between my simulation results and the paper’s results, one of which may come from the wrong coefficients I used in the lammps code. For example, the paper gives this table below
, and I need to convert the force constants of this table to the harmonic/shift bond coefficients(Umin,r0,rc) used in the lammps code. But I don’t know what the force constants given in the table refer to. Is it Kb of the potential energy expression given in the paper? Is it k of the expression “k = Umin/[2(r0 − rc)^2]” given in the lammps documentation section 5.11.3? Is it K of the expression “K=Umin/(r0 − rc)^2”? Or is it K of the expression “K = 2Umin/(r0 − rc)^2”?
The corresponding authors haven’t replied to my asking-for-help email yet.
That is not why I suggested this, but rather as a means to determine whether the parameters you use yield the desired functional form.
Given that the paper seems to provide only information about k and r0 and also since the publicly visible part of the publication only mentions using a regular harmonic potential, the inconsistency is more likely the mention of harmonic/shift. With the provided information, you can only use bond style harmonic. Apart from a constant factor in the bond energy, the result should be the same.
Since I cannot access the entire paper at the moment, could it be that the authors used the harmonic shift formulation to fit the potentials to their experimental data but then use the regular harmonic bond style with the parameters from the table for their simulations?
Thanks very much for your reply, and I apologize for not providing the full dataset and background of the paper at first which seems to confuse you about my question.
The paper does give the numeric values of r0 and rc as the below screenshot shows.
I haven’t thought about your suggested scenario before, and it does open my mind and resolve a large part of my problem. Your help is much appreciated.
Finally, regarding the k expression “k = Umin/[2(r0 − rc)^2]” in the lammps documentation section 5.11.3, it seems rather odd to me because all the other harmonic coefficients are K with the usual 1/2 factor included, and even if the k coefficient in section 5.11.3 refers to the usual spring constant of Hooke’s law, then its expression should be “k = 2Umin/(r0 − rc)^2” according to my understanding.
If you want to compute the spring constant equivalent to bond style harmonic, it would have to be K = \frac{U_{min}}{(r_0-r_c)^2}; your expression would only work if you assume \frac{dE}{dr} = \frac{1}{2}k\cdot (r-r_0)
You can confirm this using the bond_write command. Here is an an example input for that:
Looking through the source code (force calculation and parameter processing), there is in fact a typo in the documentation and I think your expression here
is correct (but see edit).
EDIT – your expression is correct for a definition of k by which
U = \frac{1}{2}k (r-r_0)^2
But in LAMMPS bond_style harmonic the coefficient K has the factor of half absorbed (presumably that is why we call it K rather than k), that is
U = K (r-r_0)^2
so for translating to bond_style harmonic coefficients you should follow @akohlmey 's post.
By the way, this is a good example of the practical value of reproducibility work. When a paper does not include LAMMPS inputs (a few kilobytes would be enough) it is difficult to reproduce, and another more easily reproducible paper will be reproduced and cited instead. It is especially absurd that the authors say “No data was used in this paper” (not even the usual “Data available upon request”).