Dear All
Now I am doing the simulation of Ga ion bombardement of Silicon.And I want to establish the model as the figure shows:
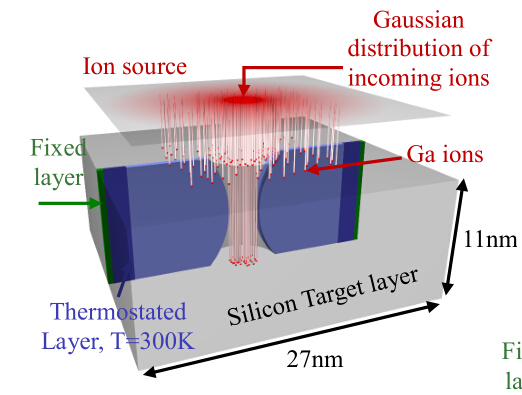
Now I have some questions in lammps command (fix deposit) to achieve this goal.
Q1:how i can guarantee the z coordinates of incident ions are the same?That is mean how I can create the ions in a circular area.I notice that if i use the command like this: region region insert sphere 135 135 120 15.0 units box; *fix 5 yinsert deposit 3 2 100 12345 region insert local 120.0 120.0 0 vz -2102.0 -2102.0 units box .*Then the initial z coordinate of all ion seems are the same ,that is 120.0,while they have different x and y coordinate.
Q2:how i can guarantee the ions are in the gaussian distribution?
Q3:I notice that in fix deposit command ,there is target keyword.The mannua says that it can make ions like a beam.So I want to make certain that the target keyword can only change the direction of the ion velocity .If i want to simulate straight beam like the figure,I just create the ions in an circular area,and give the vz velocity .Then these ions can head toward the substrate like a straight beam while ignoring target keyword .Am i right?
Best regards.
{kind=link}