I am currently using ATAT to generate SQS structures for a ceramic system, specifically a Li-rich layered oxide (LMR) type material. The cation sublattice contains multiple transition metals, and I am trying to model the disordered distribution using the mcsqs module.
I have a few questions regarding best practices for generating and evaluating SQS structures.
Number of SQS trials (parallel runs)
When generating SQS structures using mcsqs, how many independent runs are typically recommended to obtain a reliable structure?
For example, is there a common practice regarding the number of parallel attempts or different random seeds that should be tested?
Criteria for selecting the best SQS
After running mcsqs, multiple candidate structures are usually obtained (e.g., several bestsqs.out files).
What is the recommended way to determine which structure is the best SQS?
Should we mainly rely on:
the objective function value (correlation mismatch),
the bestcorr.out results,
or some other criterion?
Guidelines for ceramics / oxides
Most SQS examples I found are for metallic alloys.
Are there any specific considerations when generating SQS for ceramic systems such as oxides (e.g., layered LiMO₂ or Li₂MnO₃-based materials)?
For example:
typical supercell size
target correlation range
acceptable correlation error
If there are any recommended references, tutorials, or examples for oxide or ceramic systems, I would greatly appreciate it.
Hello,
personally, I am often using in general about 10 independent SQSs for subsequent simulations to obtain statistics. If you want the best structure from the run, then lower objective function means ‘better SQS’. I also check bestcorr.out for the best found SQS to see how it is matching with the perfect random state.
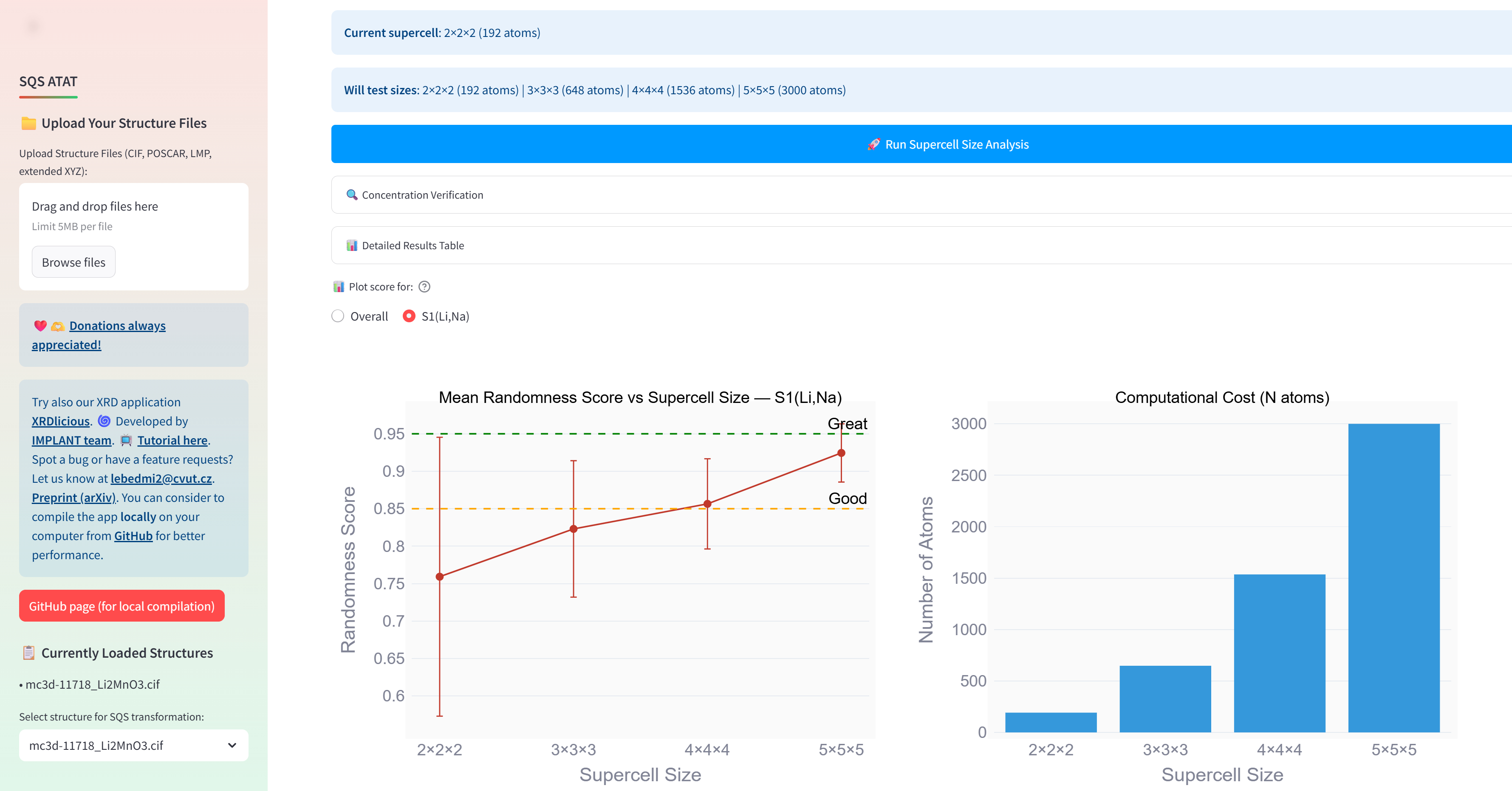
Supercell size depends on your system. For instance (see the figure below), If I use Li2MnO3 and replace Li with 75 % of Na, then if I perform analysis of randomness score based on WC vs. supercell size, I see that up to 4x4x4, it is better to use the mcsqs search to generate reasonable random structure model rather than simply generate the structure completely randomly (placing the atoms randomly on their position).
I will implement soon some general tips on https://simplysqs.com, it seems like a good idea to have it there.
I am currently practicing and trying to clarify my understanding.
In the paper, the authors performed 10 independent mcsqs runs for SrTiO₃, whereas I carried out 30 runs. As a result, I obtained lower objective function values (which is better), and at the same time, the time required for the objective function to converge became longer.
Does this mean that, although it takes more time, increasing the number of parallel runs generally allows one to obtain an SQS structure with a lower (i.e., better) objective function value? Of course, I understand that the correlation functions should still be compared explicitly.
I would like to ask one more question. In your experience, do you usually select a good SQS structure simply based on the lowest objective function value, or do you go into the bestcorr.out file and manually compare the deviations of the correlation functions from the reference random values, shell by shell (pair, triplet, etc.) before making a final decision?
Hi,
performing more parallel runs does not necessarily guarantee a better objective function value. But it increases the probability that one of the runs will land on a better configuration because of the randomness in the process.
In my experience, again, I usually keep all the SQSs from the parallel run for subsequent calculations to get statistics, but if I want just one, I will take the one with the best objective function.