I’ve finished the CE model training and new structures enumerations and predictions. Now I’d like to perform Monte Carlo simulations on my predicted structures to verify that my predicted structures are indeed the lowest energy structures at that composition.
I read from the tutorial on the website that
The canonical ensemble provides an ideal framework for studying the properties of a system at a specific concentration. Properties such as potential energy or phenomena such as chemical ordering at a specific temperature can conveniently be studied by simulating at that temperature. The canonical ensemble is also a convenient tool for “optimizing” a system, i.e., finding its lowest energy chemical ordering. In practice, this is usually achieved by simulated annealing, i.e., the system is equilibrated at a high temperature, after which the temperature is continuously lowered until the acceptance probability is almost zero. In a well-behaved system, the chemical ordering at that point corresponds to a low-energy structure, possibly the global minimum at that particular concentration.
However, I’m still unsure how to proceed.
I have predicted ~500,000 structures with a well‑trained CE model, computed their mixing energies, and identified 11 ground‑state structures at different compositions. I would now like to verify—via Monte Carlo at finite temperatures—that each of these 11 structures is indeed the lowest‑energy state at its composition. My specific questions are:
In a canonical‑ensemble workflow, should I initialize the ClusterExpansionCalculator with each ground‑state structure and then pass it to CanonicalEnsemble() for the MC run?
Should I perform separate MC simulations for each ground‑state composition? Or it’s possible to do it together?
After the MC runs, how should I analyze the results to confirm whether these structures are the lowest‑energy states at their respective compositions? As indicated in the tutorial, The canonical ensemble is also a convenient tool for “optimizing” a system, i.e., finding its lowest energy chemical ordering.
I’m sorry that I feel confused at this moment. I would really appreciate it if you can help explain it.
Yes you can do it like this. The structure you pass to the Ensemble will be the initial structure in the MC simulation (see the docs for this Ensembles — icet documentation)
It sounds like you want to do annealing (cooling) simulations to find the ground-state, then you need to run one such simulation for each composition you are interested in (11?)
You obtain the energies and structures along the simulation, so you can plot energy vs MC-step (or temperature) and see how it looks. You can extract the structure with lowest energy from the simulation and compare it to your possible candidate ground-state from your prior analysis.
Thank you so much for the reply. I really appreciate it. I still have some confusion while trying it out.
1) Initialization / search space
When initializing ClusterExpansionCalculator, should I supply only my predicted ground‑state structure, or first use generate_sqs() to create multiple initial configurations?
If I provide a single structure, will the subsequent canonical‑ensemble MC explore only site swaps within that fixed lattice, composition, and cell (i.e., not sample other orderings unless I supply them explicitly)? I’d appreciate clarification of the search space implied by the initialization.
2) Representing vacancies (NaCl‑type, two sublattices; N‑sublattice has vacancies)
In ICET 2.2 I trained with chemical_symbols=[['Ti','V'], ['N','X']], where X denotes vacancies. The newest version requires each symbol to be a real element, and ASE/pymatgen do not automatically interpret X or Va as vacancies.
Would using a placeholder element (e.g., H) to stand in for vacancies be acceptable for generating events and running MC, or would that introduce artifacts?
Is there a recommended workflow for labeling and handling vacancies across ICET/ASE without resorting to a fictitious element?
Thank you so much for your time and attention. Look forward to your reply.
It ends up with error: ValueError: Occupations of structure not compatible with the sublattice. Site 5 with occupation X not allowed on sublattice (‘H’, ‘N’)
So it seems that ICET 2.2 identify vacancies with X and doesn’t allow for using other placeholders. Do you think it’s gonna work if I just don’t do anyting on the vacancy labeling and leave it in the form in POSCAR and feed it directly to MC run? Do you think MC run will expect me to labelize the vacancies?
Yes you can only supply it with one structure (see the documentation page for ClusterExpansionCalculator. This calculator will work for any occupation/composition of this given supercell.
In the canonical ensemble you’re concentrations (number of atoms of each species) is fixed by definition. I think you’d want to use the CanonicalAnnealing ensemble. The canonical ensemble will search for any ordering or occupation in this supercell which has this given composition. Meaning it can swap position of two atoms, but it can not change atom of type A into atom of type B.
Yes using any placeholder element to represent vacancies works fine (as long as this element is not present elsewhere in the system ofcourse).
No I dont think there is a better way to represent vacancies, placeholder atoms is the way to do it I think.
In your code example, you must have some elements in slab that are represented as ‘X’?
ICET 2.2 or newest version doesnt know about vacancies, so you must treat them as any other atom type.
No I dont think your suggestion would work, you need to use placeholder atom to represent vacancies.
sc = StructureContainer(cluster_space=cs)
for row in db.select():
sc.add_structure(structure=row.toatoms(),
properties={‘mixing_energy’: row.mixing_energy})
Here, I use “H” as the placeholder for vacancy, but it ends up with error: ValueError: Occupations of structure not compatible with the sublattice. Site 5 with occupation X not allowed on sublattice (‘H’, ‘N’)
I think when I mapped the structures into ideal structures, ICET has automatically labelled the vacancies as “X”, thus doesn’t allow me to use “H” as placeholder. But when I run with
I don’t think I set vacancies as X explicitly, I believe it’s set automatically by map_structure_to_reference() when I map the structures to primitive structures. I’m not sure how I can rename the X to H, should I modify the ase.db file that contains the structure information?
I have just tried to map my predicted structures into a reference structure by using map_structure_to_reference() and write it into a ase.json file that can be read directly by ICET when doing MC run. And it works well. Here attached the json file that I’d like to use for MC run. In this file, by using map_structure_to_reference(), it recognizes the vacancies and identify them with “X”. Do you think I can use this structure directly for MC run without explicitly using other placeholders for vacancies?
Yes maybe my confusion comes from that why cant you just swap atoms denoted ‘X’ to atoms denoted by ‘H’ (if using ‘X’ to denote vacancies doesnt work), e.g.
atoms.symbols[atoms.symbols=='X'] = 'H'
If you do this before writing you structures to a database, or after reading them and before handing them to the structure container doesnt matter.
ICET doesnt read json files, thats ASE. If using databases complicates matters then no need to use them.
ICET just requires ASE-Atoms objects.
Thank you so much for the clarification. It’s really helpful.
I quite agree with your proposed method, it’s indeed simpler and more straightforward. I have complicated my workflow at first and it’s kind of hard to readjust now.
I have enumerated and relaxed around 5000 structures and then calculated their formation energies. Then I use the following commands
to read from each subdirectories and obtain their structure and energy information. But here, pymatgen and ASE atoms cannot identify vacancies in the structures automatically, so I have to map these structures back to ideal structures to obtain the structure information with vacancies, denoted by “X” by map_structure_to_reference(). And CE training also requires ideal structures.
I guess I after obtaining the ideal structures, I should directly use the way you mentioned, like
Also, if I got the warning that The ClusterExpansionCalculator self-interacts, which may lead to erroneous results. To avoid self-interaction, use a larger supercell or a cluster space with shorter cutoffs.
Does it affect the results if I expand the cell? Since the prediction didn’t go up to such a large cell.
After running Monte Carlo starting from my predicted structure at 1000 K and 300 K for 5×10⁵ steps on a much larger supercell, a lower‑energy configuration emerged with ΔE ≈ 10 meV/site. I’m cautious about this result because the larger ground state may have a longer periodicity than my original enumeration allowed; thus, the smaller‑cell enumeration cannot guarantee optimality in the expanded cell.
Given a CE MAE of ~9 meV/site, does a 10 meV/site difference meaningfully challenge the earlier ground‑state prediction, or is it within expected error?
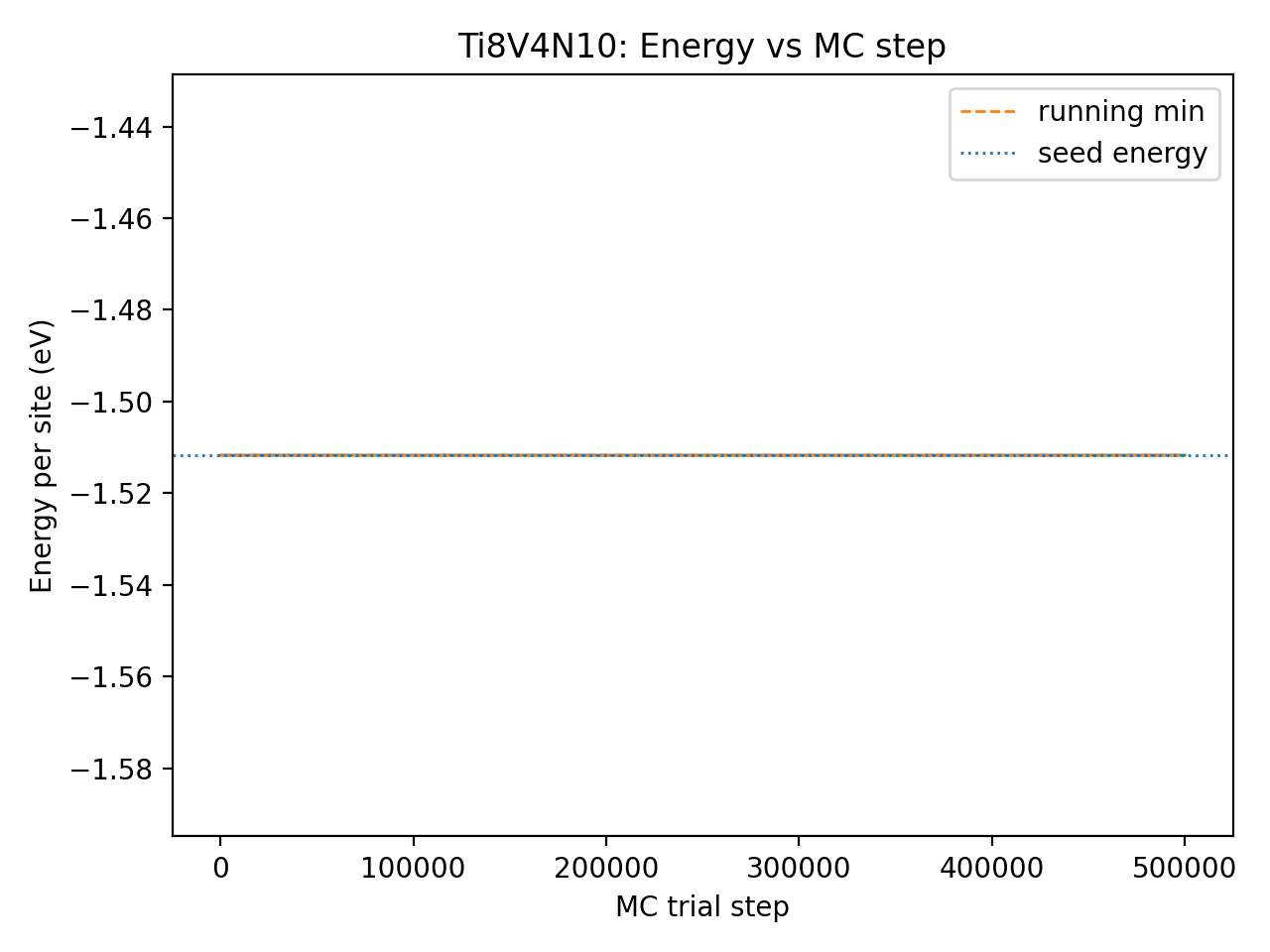
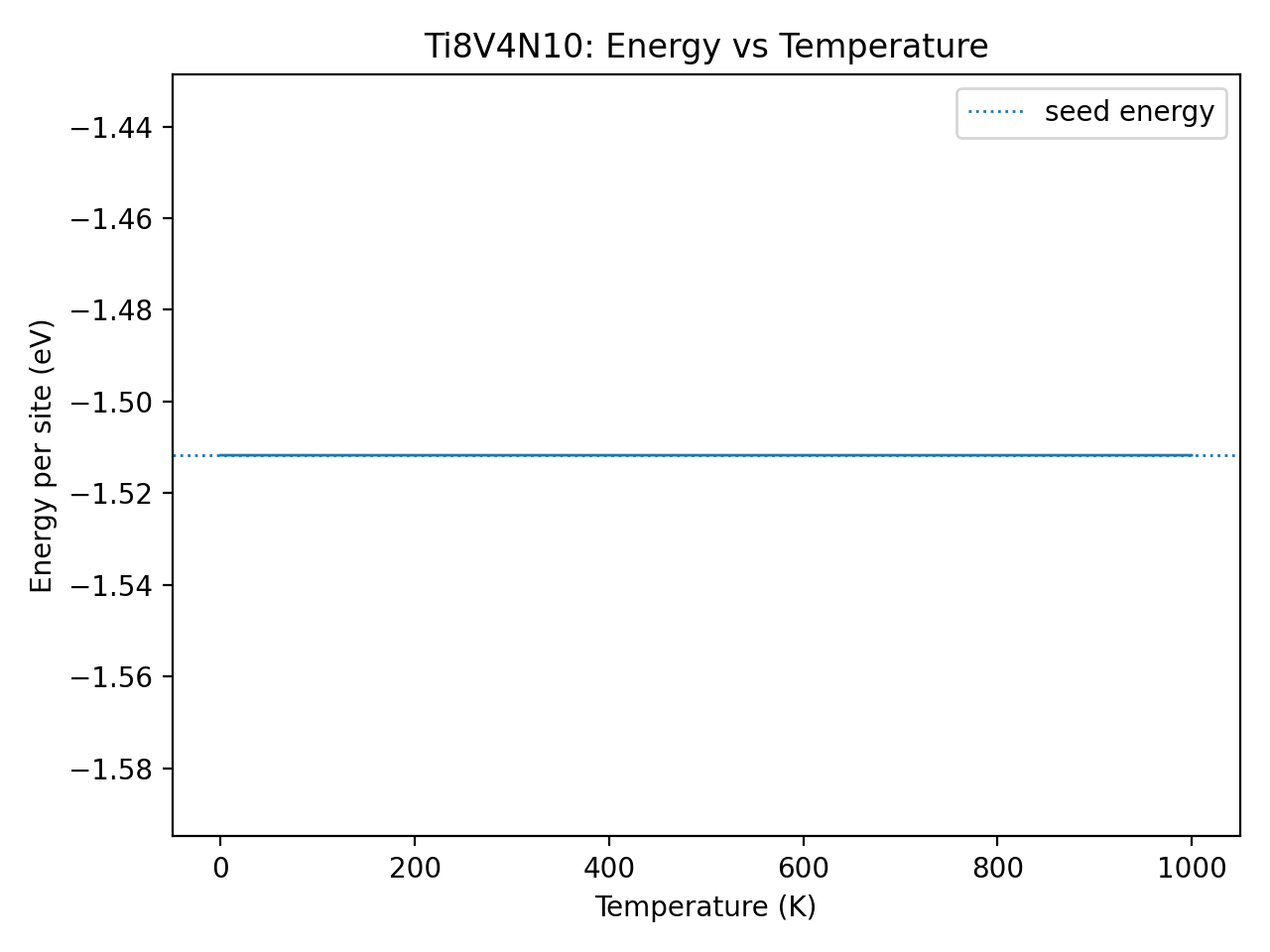
When I run MC on the unexpanded 24‑site cell, ΔE_eV_per_site stays at 0 and the energy–temperature curve is flat, suggesting no effective sampling (e.g., no accepted moves). How can I configure MC on the original cell to explore configurations properly and provide a meaningful validation?
If you found a structure that is 10meV/site lower than your prior ground-state and your error is about 9meV/site, then it is likely but not certain that this is actually lower in energy.
Only way to know for sure would be to check the relaxed energies of the two structures with DFT.
You run a canonicalAnnealing simulation as written above, where you start from high temperature where moves will be accepted
Yes, that’s exactly what I did. But the tricky part is the cell size. My original predicted structure has only 24 atoms, but when I feed this cell to MC, it says
Therefore, I have to expand the cell. But I want to try my original smaller cell (with 24 atoms). If I use the smaller cell, the self-interacts warning somehow prevents the algorithm from doing anything, and the structure/energy stay exactly the same. As attached in the figure.
This is not true, the warning is just telling you that the cell is small compared to the cutoffs.
I think you need to do some trial and error here. Have you tried increasing temperature to 10,000 or 100,000 still nothing changes?
If so, are you sure the structure you run with allows for any swaps?
I haven’t tried such high temperature, I only tried 1000 K, but since the larger cell will change at 1000 K, I think 1000 K should be high enough for it to undergo changes.
Actually, this is the question I wanna ask. I think the structure didn’t go through any swaps. So how can I confirm if the structure allows for swaps and how can I make it allow for swap? I didn’t do anything to prevent it from swaps, I just use the basic settings. Here is my snippet.
try:
mc = CanonicalAnnealing(
structure=structure,
calculator=calc,
T_start=T_start,
T_stop=T_stop,
n_steps=int(n_steps),
user_tag=tag,
random_seed=seed,
dc_filename=None,
trajectory_write_interval=None,
ensemble_data_write_interval=int(write_interval),
cooling_function="exponential",
)
except TypeError:
# Some builds may not support all kwargs; fall back to essentials.
mc = CanonicalAnnealing(
structure=structure,
calculator=calc,
T_start=T_start,
T_stop=T_stop,
n_steps=int(n_steps),
user_tag=tag,
random_seed=seed,
)
mc.run()
No thats not necessarily the case, different supercell sizes behave differently,
I think you should try higher temperatures if you want to resolve this.
Also try cooling function linear rather than exponential.
The resulting datacontainer from an MC run contains the number of accepted trial swaps that you can check
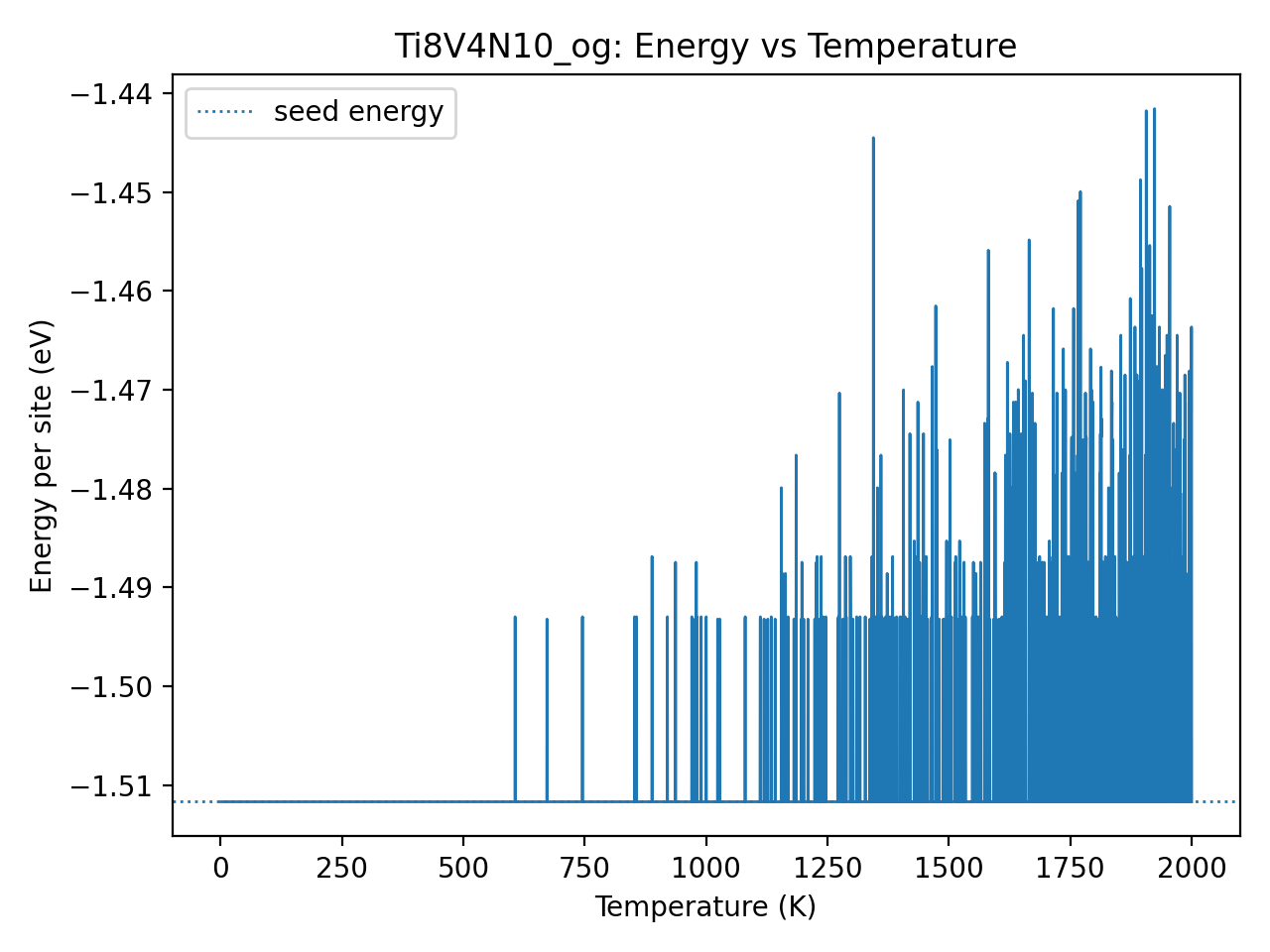
I have tried higher temperatures (i.e., 2000 K) for my initial structure (24 atoms) with linear cooling function. The attached figures show the E vs T and E vs steps, now it’s not a flat line.
However, when I checked the aceepted trial swaps in datacontainer by using the following commands
Is it correct to calculate the swap number this way? The number of swaps is 61616, which seems reasonable. Since the swap count is non-zero, can we infer that the algorithm has explored a significant number of alternative chemical orderings before confirming our current structure as the most stable one?
Also, if you agree that the energy plot looks good, would it be fair to conclude that the predicted structure likely represents the lowest-energy configuration for this composition and cell size? Based on the current evidence, do you think it’s appropriate to make this claim?