Dear LAMMPS developers,
I am running the simulation for a system with flexible spc and flexible co2, and silica surfaces. I found the different pressures in the bulk water region between the Verlet integrator and r-RESPA integrator. I used a timestep of 0.25 fs for Verlet and the following set up for r-RESPA:
run_style respa 3 4 2 bond 1 angle 1 dihedral 1 improper 1 pair 2 kspace 3,
so that the bond and angle forces are calculated every 0.25 fs, pair forces at 1 fs and kspace at 2 fs. For kspace calculation (pair_style lj/cut/coul/long), pppm with accuracy 1e-6 was used.
The pressure in bulk water region (far away from the interfaces) with Verlet is around 12 MPa, while one with r-RESPA gives 115 MPa, which is 10x larger. The pressures in bulk co2 region from the two systems are statistically similar. I am aware when using large timestep for flexible water with Verlet integrator, the calculation of pressure would be incorrect, as I have tested something similar in http://lammps.sandia.gov/threads/msg66029.html (Although I got correct pressures for rigid spc at 1 fs…). So, would this means that the way of the pressure calculation in r-RESPA (at a larger timestep?) cause the problem?
Thank you for your time.
Regards,
Pengyu
Just forgot to mention in my previous email, the outermost timestep is 2 fs in r-RESPA, so that the bond and angle forces are calculated every 0.25 fs, pair forces at 1 fs and kspace at 2 fs.
Regards,
Pengyu
you also forgot to mention the LAMMPS version you are using and
whether you get the same issue, when you run r-RESPA with 0.25fs time
step and only one level or a multiplier of 1 (so that all levels run
with the same size timestep).
finally, you mention a multi-component system so, you should make
tests with a homogeneous system first.
axel.
Thank you, Axel, for your reply.
Sorry that I should have provided more details and further tests. The LAMMPS version I am using is the previous stable version 11Aug2017. I also did a quick check, it seems like the r-RESPA has not been changed.
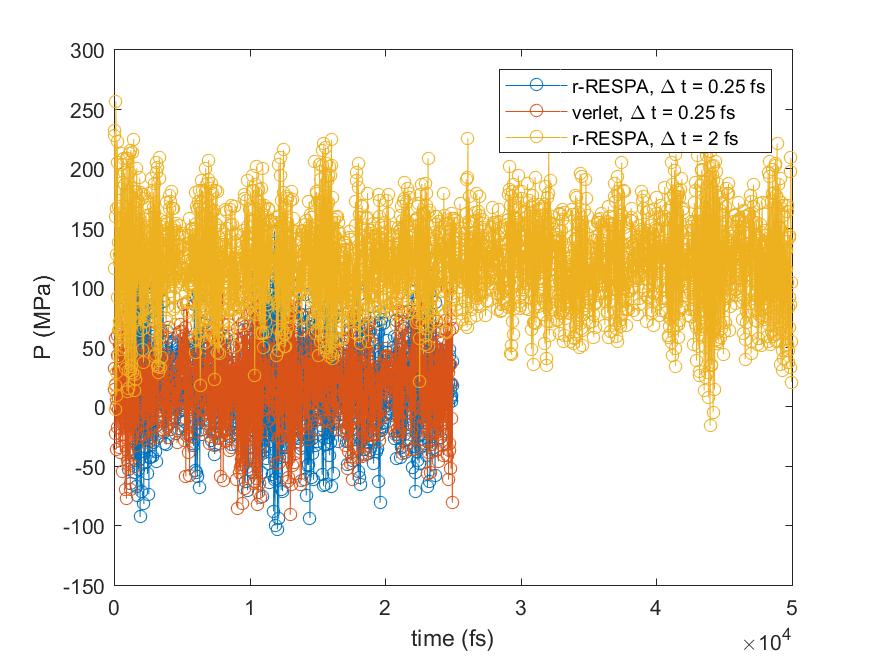
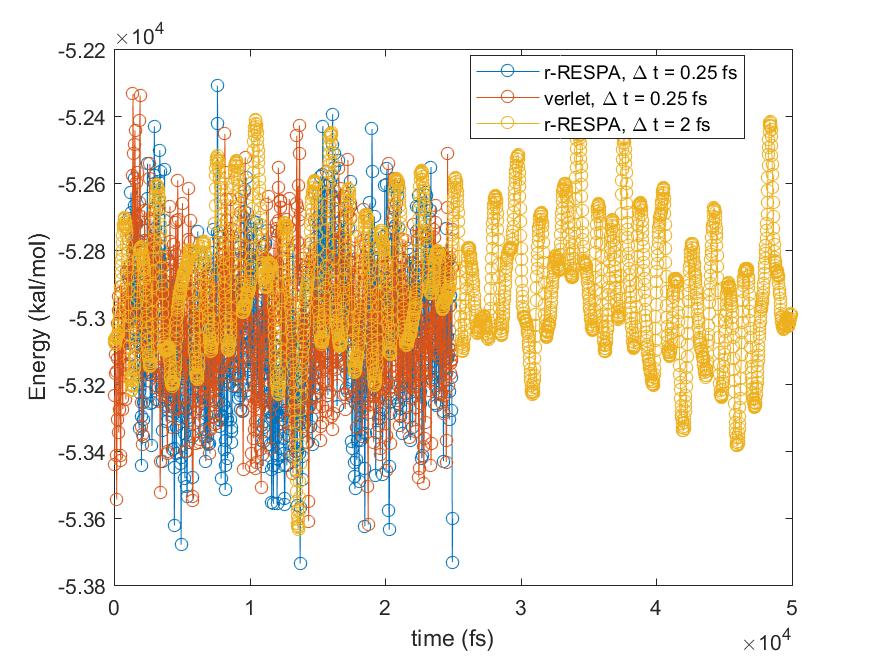
I did a quick test with different integrators for the flexible SPC running in the NVT ensemble with “fix nvt” at T = 333.15K, restarting from a pre-equilibrated water box (which has 8287 SPC molecules) in the NPT ensemble at a targeted pressure of 20 MPa and T = 333.15 K. I have compared three integration methods: 1. r-RESPA with timestep = 0.25 fs and only one level for all forces calculations; 2 Verlet with timestep = 0.25 fs; 3 r-RESPA with timestep = 2 fs, with bond and angles calculated at 0.25 fs, pair at 1 fs and kspace at 2 fs (run_style respa 3 4 2 bond 1 angle 1 dihedral 1 improper 1 pair 2 kspace 3). In all cases, the Tdamp was 100 times of the timestep (or outermost timestep).
I have attached the plots for the total energy and the pressure against with time. The simulation was run 25 ps for the Cases 1 and 2, and 50 ps for the Case 3. It may be a bit short, but I think it already captures the problem I described. The Case 1 and 2 had similar results, while the Case 3 had higher total energy and pressure than the Cases 1 and 2. The pressure for Cases 1 and 2 were around 16 -18 MPa, respectively, while the Case 3 had an average pressure of 120 MPa. All cases had similar degrees of fluctuations, given the standard deviations were around 35-40 MPa. If there is no problem with the r-RESPA, would this means that I cannot use it for flexible water to get the correct pressure?
Regards,
Pengyu
Thank you, Axel, for your reply.
Sorry that I should have provided more details and further tests. The LAMMPS
version I am using is the previous stable version 11Aug2017. I also did a
quick check, it seems like the r-RESPA has not been changed.
that is not so easy to say. the respa.cpp and respa.h files only
contain the scheduling of the calculations. there are plenty of RESPA
specific functions scattered throughout other source files, which can
have an impact on the output.
I did a quick test with different integrators for the flexible SPC running
in the NVT ensemble with "fix nvt" at T = 333.15K, restarting from a
pre-equilibrated water box (which has 8287 SPC molecules) in the NPT
ensemble at a targeted pressure of 20 MPa and T = 333.15 K. I have compared
three integration methods: 1. r-RESPA with timestep = 0.25 fs and only one
level for all forces calculations; 2 Verlet with timestep = 0.25 fs; 3
r-RESPA with timestep = 2 fs, with bond and angles calculated at 0.25 fs,
pair at 1 fs and kspace at 2 fs (run_style respa 3 4 2 bond 1 angle 1
dihedral 1 improper 1 pair 2 kspace 3). In all cases, the Tdamp was 100
times of the timestep (or outermost timestep).
I have attached the plots for the total energy and the pressure against with
time. The simulation was run 25 ps for the Cases 1 and 2, and 50 ps for the
Case 3. It may be a bit short, but I think it already captures the problem I
described. The Case 1 and 2 had similar results, while the Case 3 had higher
total energy and pressure than the Cases 1 and 2. The pressure for Cases 1
and 2 were around 16 -18 MPa, respectively, while the Case 3 had an average
pressure of 120 MPa. All cases had similar degrees of fluctuations, given
the standard deviations were around 35-40 MPa. If there is no problem with
the r-RESPA, would this means that I cannot use it for flexible water to get
the correct pressure?
so far you have confirmed, that the r-RESPA formulation in LAMMPS
falls back on the verlet code even when using the various levels to
compute forces and stress.
the next step would be to determine systematically, where the pressure
shift is coming from and whether it is dependent on the timestep.
i suggest you start with using an outer time step of 0.5 fs and
run_style respa 2 2 bond 1 angle 1 dihedral 1 improper 1 pair 1 kspace 2
as well as with
run_style respa 2 2 bond 1 angle 1 dihedral 1 improper 1 pair 2 kspace 2
then time step of 1fs with respa 2 4, then time step 2fs with respa 2 8
this should allow you to identify whether the pressure shift is
dependent on either pair or kspace or both and whether it happens
suddenly or gradually.
axel.
Thanks for your suggestion, Axel.
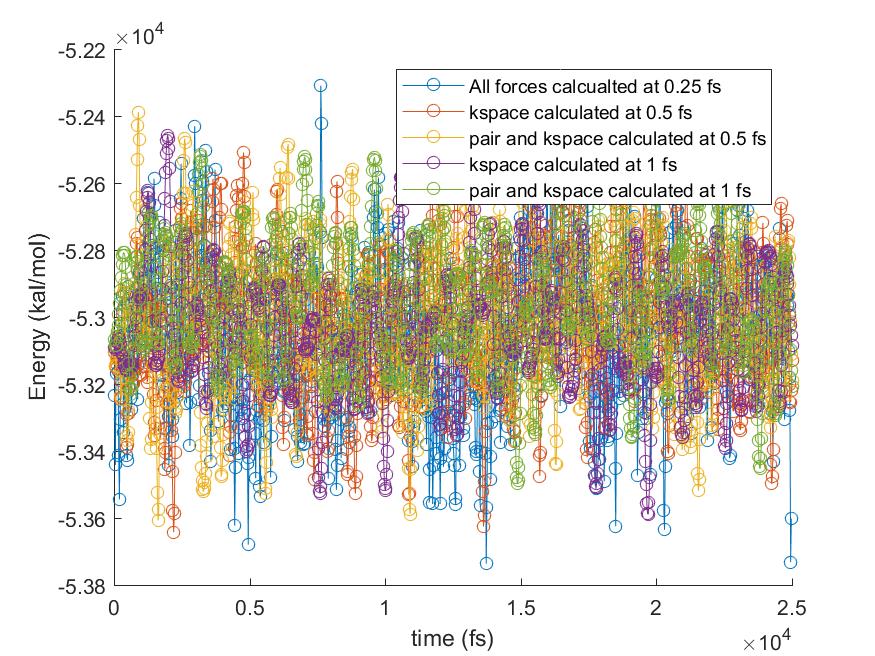
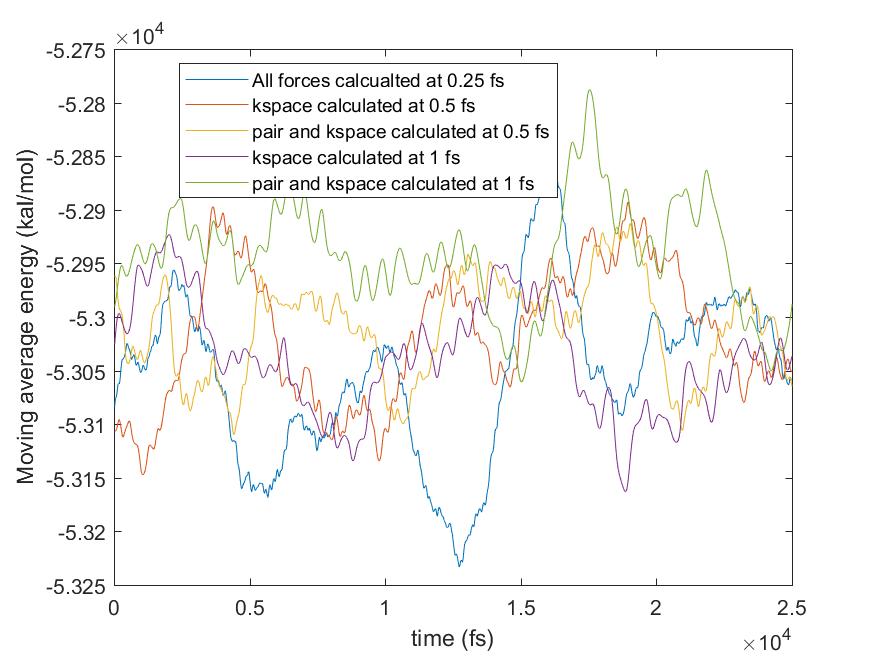
Please find attached the results of total energy and pressure against time from more tests done with r-RESPA.
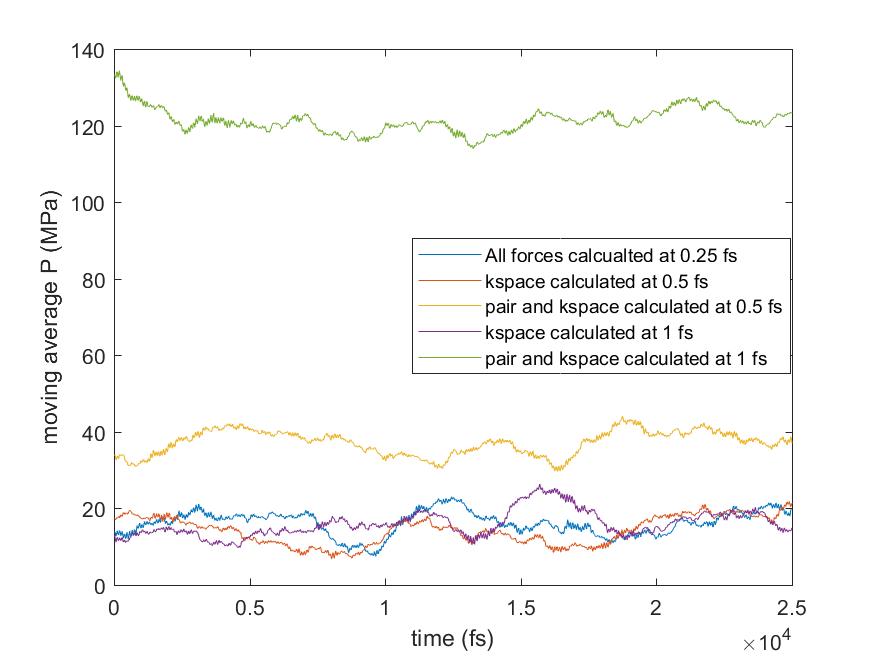
All bond and angles were calculated at every 0.25 fs and only outermost timestep was varied up to 1 fs for the pair and kspace calculation, as you suggested (I did not try 2 fs though). The results showed that the pressure shift was from the pair forces calculation with larger timesteps. The moving averages are also plotted so that it is easier to compare the changes in pressure. What do you think about the results? Would it be any problem with r-RESPA formulation in LAMMPS? Or simply we cannot simulate the flexible water with a larger timestep for correct pressure calculation?
Regards,
Pengyu
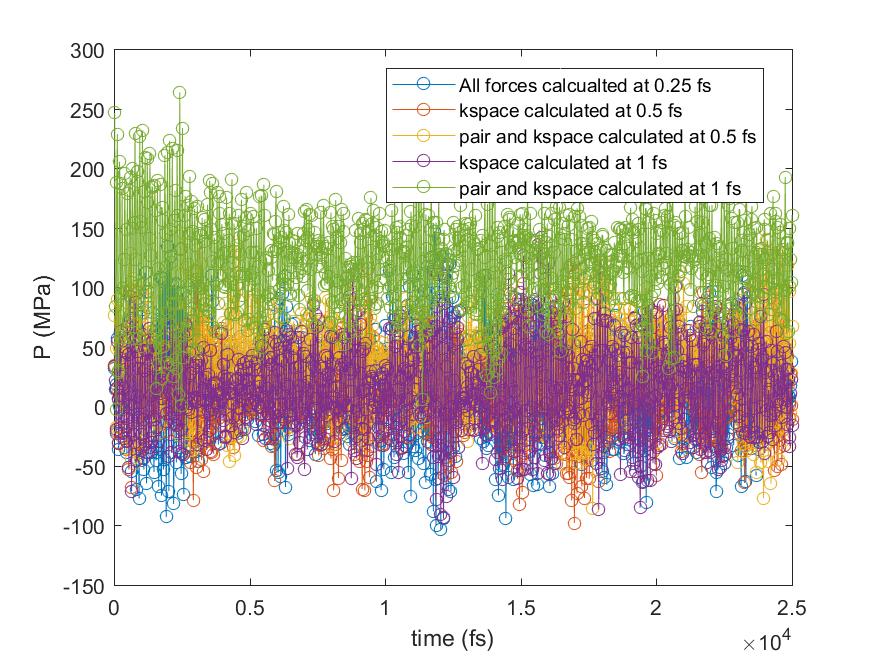
Thanks for your suggestion, Axel.
Please find attached the results of total energy and pressure against time
from more tests done with r-RESPA.
All bond and angles were calculated at every 0.25 fs and only outermost
timestep was varied up to 1 fs for the pair and kspace calculation, as you
suggested (I did not try 2 fs though). The results showed that the pressure
shift was from the pair forces calculation with larger timesteps. The moving
averages are also plotted so that it is easier to compare the changes in
pressure. What do you think about the results? Would it be any problem with
r-RESPA formulation in LAMMPS? Or simply we cannot simulate the flexible
water with a larger timestep for correct pressure calculation?
it is not my job to make your conclusions for you.
since you can single out the pairwise interactions there may be other
factors related to those, that you have not checked out yet, and that
may have an impact here. for example, the neighborlist update settings
or the cutoff(s).
axel.
Thanks Axel.
I did not see any dangerous builds from those tests so I think it may be unlikely due to the neighbor list update. Anyway, I will have a look at the code when I have time and see if I can figure out myself. Thanks for your help again.
Regards,
Pengyu
Hi Axel,
Sorry to bother you again. I have found a paper about pressure calculation for r-REPSA method: https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.24731. It suggests that the pressure calculation should include the contribution from the forces calculated at the intermediate timestep as well (between innermost and outermost timesteps). It seems like the compute pressure command only calculated with the forces at every outermost timestep (correct me if I am wrong). I am not sure whether the method of pressure calculation they proposed would resolve the problem. If I would like to implement that method in LAMMPS, would it be lots of work? since I have little experience with c++.
Thank you for your time.
Regards,
Pengyu