Hi Lammps users,
I want to calculate RDF of my solute. It is a solute solvent mixture.
How can we calculate RDF at larger distances?
(1) In which part of the program should we type rerun command?
(2) How to post process dump files?
(3) Since my solute is an organic molecule, it is numbered as C1, C2 and so on (The atoms are numbered using Gaussian software.)
How can we compute rdf of each and every atom? What command should we use?
Thanks,
Kulkarni.R
dimension 3
Use rerun in place of run. It reads the dump files. To compute
the RDF at longer distances, use a longer cutoff on the potential.
You do not need a fancy potential or any bond.angle,etc terms.
Just a simple pair lj/cut will do.
Steve
Thank you for the reply.
I am able to get RDF at longer distances.
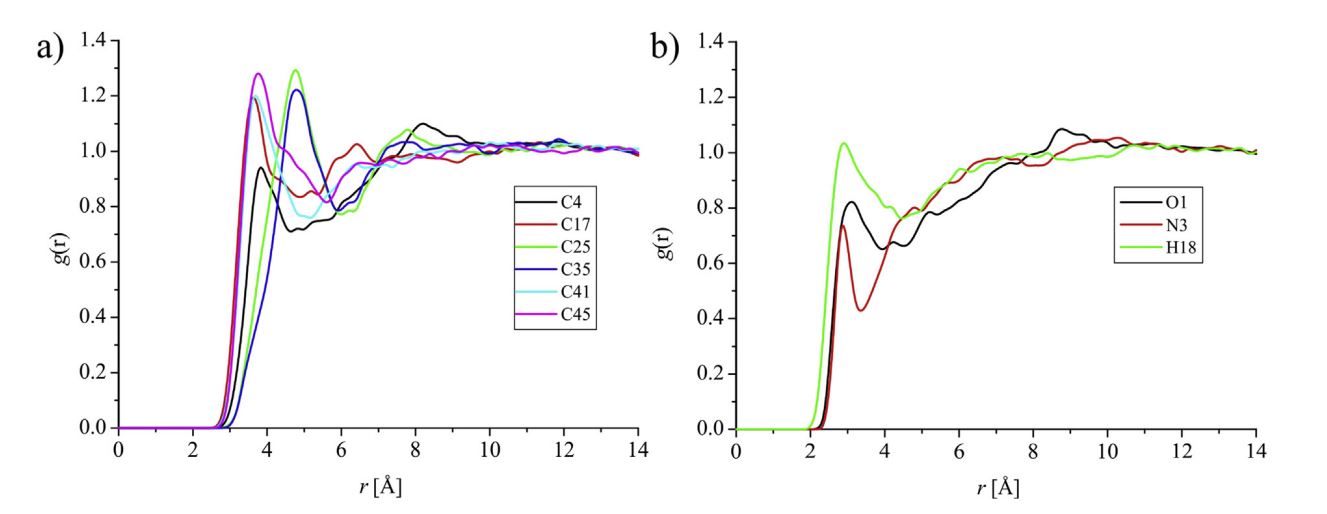
While reading papers related to RDF, came across this graph.
The authors have plotted RDF for each and every atom of their organic compound
using schrodinger materials science suite.
What would be the corresponding command in Lammps?
Thanks,
Kulkarni.R
RDF is a pairwise metric, so not sure what you mean.
If you mean a non-periodic system with N atoms,
and you want the RDF for all N^2 pairs, then
you just need to use a long enough cutoff, i.e.
the diameter of the system. In a periodic box,
you can use as large a cutoff as you want, you
will get RDF pairs with all the image atoms.
Steve
Thank you for the reply.
I am able to get RDF at longer distances.
While reading papers related to RDF, came across this graph.
The authors have plotted RDF for each and every atom of their organic
compound
using schrodinger materials science suite.
What would be the corresponding command in Lammps?
you need to assign a different atom type to each atom in the compound.
compute rdf discriminates atoms by atom type.
axel